当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Atomistic Origins of Enhanced Band Gap, Miscibility, and Oxidation Resistance in α-CsPb1–xSnxI3 Mixed Perovskite
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2020-11-19 , DOI: 10.1021/acs.jpcc.0c07356 Fernando Valadares 1 , Ivan Guilhon 1 , Lara K. Teles 1 , Marcelo Marques 1
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2020-11-19 , DOI: 10.1021/acs.jpcc.0c07356 Fernando Valadares 1 , Ivan Guilhon 1 , Lara K. Teles 1 , Marcelo Marques 1
Affiliation
|
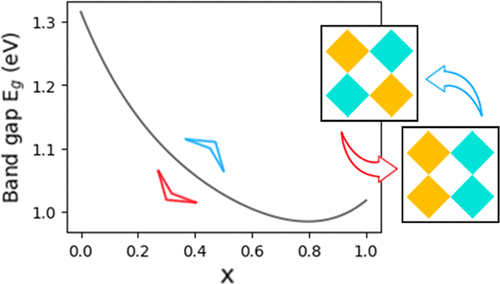
The advance made in perovskite-based solar cell technology demands the search for materials with better properties, namely, high stability in operational conditions and suitable electronic structure parameters. In this work, we provide a detailed study for cubic CsPb1–xSnxI3 alloys. We employed a theoretical model that combines quasiparticle effects via the density functional theory (DFT)-1/2 method and spin–orbit corrections with a rigorous statistical disordered description of the alloy. As the main result, a reliable quantitative expression for the variation of energy gap with the composition that can be directly compared with experiments is given. A particular situation is verified for x = 0.80, where the alloy is predicted to have a band gap of 0.984, 0.033 eV lower than the gap of CsSnI3. Additionally, the model encompasses the nuances necessary to understand the properties’ behavior in this complex system. We verified that “more mixed” configurations are energetically favored, leading to ordering in very small temperatures that evolve to a very stable alloy in the usual growth conditions. Also, based on the thermodynamic results, an antioxidant mechanism for this alloy is proposed. The bowing mechanism is explained in terms of band character and spin–orbit interaction. Finally, the conclusions support CsPb1–xSnxI3 alloys as a very good potential candidate for photovoltaic applications.
中文翻译:

在α-CSPB增强带隙,可混合性,和耐氧化性的原子论起源1- X Sn的X我3混合钙钛矿
基于钙钛矿的太阳能电池技术的进步要求寻找具有更好性能的材料,即在操作条件下具有较高的稳定性以及合适的电子结构参数。在这项工作中,我们提供了对立方CsPb 1– x Sn x I 3合金的详细研究。我们采用了一种理论模型,该模型结合了通过密度泛函理论(DFT)-1/2方法的准粒子效应和自旋轨道校正以及对合金的严格统计无序描述。作为主要结果,给出了能隙随组成的变化的可靠定量表达式,可以直接与实验进行比较。针对x验证了一种特殊情况= 0.80,其中预测该合金的带隙为0.984,比CsSnI 3的带隙低0.033eV 。此外,该模型还包含了解该复杂系统中的属性行为所必需的细微差别。我们验证了在能量上偏爱“更多混合”的构型,从而导致在非常小的温度下有序化,而在通常的生长条件下,这种温度会演变成非常稳定的合金。另外,基于热力学结果,提出了该合金的抗氧化机理。弯曲机制是根据波段特征和自旋轨道相互作用来解释的。最后,结论支持CsPb 1– x Sn x I 3合金是光伏应用中非常好的潜在候选材料。
更新日期:2020-12-03
中文翻译:
在α-CSPB增强带隙,可混合性,和耐氧化性的原子论起源1- X Sn的X我3混合钙钛矿
基于钙钛矿的太阳能电池技术的进步要求寻找具有更好性能的材料,即在操作条件下具有较高的稳定性以及合适的电子结构参数。在这项工作中,我们提供了对立方CsPb 1– x Sn x I 3合金的详细研究。我们采用了一种理论模型,该模型结合了通过密度泛函理论(DFT)-1/2方法的准粒子效应和自旋轨道校正以及对合金的严格统计无序描述。作为主要结果,给出了能隙随组成的变化的可靠定量表达式,可以直接与实验进行比较。针对x验证了一种特殊情况= 0.80,其中预测该合金的带隙为0.984,比CsSnI 3的带隙低0.033eV 。此外,该模型还包含了解该复杂系统中的属性行为所必需的细微差别。我们验证了在能量上偏爱“更多混合”的构型,从而导致在非常小的温度下有序化,而在通常的生长条件下,这种温度会演变成非常稳定的合金。另外,基于热力学结果,提出了该合金的抗氧化机理。弯曲机制是根据波段特征和自旋轨道相互作用来解释的。最后,结论支持CsPb 1– x Sn x I 3合金是光伏应用中非常好的潜在候选材料。










































 京公网安备 11010802027423号
京公网安备 11010802027423号