当前位置:
X-MOL 学术
›
Comp. Mater. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Cu nanoprecipitate morphologies and interfacial energy densities in bcc Fe from density functional theory (DFT)
Computational Materials Science ( IF 3.1 ) Pub Date : 2021-02-01 , DOI: 10.1016/j.commatsci.2020.110149 A.M. Garrett , C.P. Race
Computational Materials Science ( IF 3.1 ) Pub Date : 2021-02-01 , DOI: 10.1016/j.commatsci.2020.110149 A.M. Garrett , C.P. Race
|
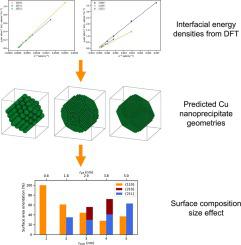
Abstract We use quantum mechanical ab initio simulation to inform a model which predicts the structures of coherent Cu nanoprecipitates that are thought to form in low-alloy reactor pressure vessel (RPV) steels. Using density functional theory (DFT) we calculated the interfacial energy densities of {1 0 0}, {1 1 0}, {1 1 1}, {2 1 0}, {2 1 1} and {2 2 1} orientated Fe-Cu interfaces. These energy density values were used in an optimisation model to predict low energy Cu nanoprecipitate geometries for nominal radii ranging from 1 to 5 nm. Strain states parallel to the Fe-Cu interface were calculated using an embedded atom method (EAM) potential for similarly sized Cu nanoprecipitates. Fe-Cu interfacial energy densities under equivalent strains were calculated using DFT allowing comparison with the predictions of the EAM potential. The DFT calculations revealed the lowest energy Fe-Cu interface orientation to be the {1 1 0}. Accordingly, the geometry prediction optimisations found that regardless of size the predicted Cu nanoprecipitate surface geometries were dominated by the {1 1 0} orientation. Notably, as the Cu nanoprecipitates increased in size the ratio of the surface made up of non-{1 1 0} orientations was observed to proportionally increase. This allows the nanoprecipitate to take a more spherical form so reducing its total surface area. Additionally, the Fe-Cu interface strains predicted by the EAM potential for all Cu nanoprecipitate radii are sufficiently small that they do not significantly alter the calculated interfacial energy density values. This finding suggests interfacial strain does not play a significant role in determining the morphology of Cu nanoprecipitates.
中文翻译:

来自密度泛函理论 (DFT) 的体心立方 Fe 中的 Cu 纳米沉淀物形态和界面能量密度
摘要 我们使用量子力学 ab initio 模拟为模型提供信息,该模型预测被认为在低合金反应堆压力容器 (RPV) 钢中形成的相干 Cu 纳米沉淀物的结构。使用密度泛函理论 (DFT),我们计算了 {1 0 0}、{1 1 0}、{1 1 1}、{2 1 0}、{2 1 1} 和 {2 2 1} 取向的界面能量密度Fe-Cu 界面。这些能量密度值用于优化模型,以预测标称半径范围为 1 至 5 nm 的低能量 Cu 纳米沉淀物的几何形状。平行于 Fe-Cu 界面的应变状态是使用嵌入原子法 (EAM) 计算出类似尺寸的 Cu 纳米沉淀物的电位。使用 DFT 计算等效应变下的 Fe-Cu 界面能量密度,允许与 EAM 潜力的预测进行比较。DFT 计算显示能量最低的 Fe-Cu 界面取向为 {1 1 0}。因此,几何预测优化发现,无论大小,预测的 Cu 纳米沉淀表面几何形状都受 {1 1 0} 取向支配。值得注意的是,随着Cu纳米沉淀物尺寸的增加,观察到由非{1 1 0}取向构成的表面比例成比例地增加。这允许纳米沉淀物采取更球形的形式,从而减少其总表面积。此外,所有铜纳米沉淀半径的 EAM 电位预测的 Fe-Cu 界面应变足够小,它们不会显着改变计算的界面能量密度值。
更新日期:2021-02-01
中文翻译:
来自密度泛函理论 (DFT) 的体心立方 Fe 中的 Cu 纳米沉淀物形态和界面能量密度
摘要 我们使用量子力学 ab initio 模拟为模型提供信息,该模型预测被认为在低合金反应堆压力容器 (RPV) 钢中形成的相干 Cu 纳米沉淀物的结构。使用密度泛函理论 (DFT),我们计算了 {1 0 0}、{1 1 0}、{1 1 1}、{2 1 0}、{2 1 1} 和 {2 2 1} 取向的界面能量密度Fe-Cu 界面。这些能量密度值用于优化模型,以预测标称半径范围为 1 至 5 nm 的低能量 Cu 纳米沉淀物的几何形状。平行于 Fe-Cu 界面的应变状态是使用嵌入原子法 (EAM) 计算出类似尺寸的 Cu 纳米沉淀物的电位。使用 DFT 计算等效应变下的 Fe-Cu 界面能量密度,允许与 EAM 潜力的预测进行比较。DFT 计算显示能量最低的 Fe-Cu 界面取向为 {1 1 0}。因此,几何预测优化发现,无论大小,预测的 Cu 纳米沉淀表面几何形状都受 {1 1 0} 取向支配。值得注意的是,随着Cu纳米沉淀物尺寸的增加,观察到由非{1 1 0}取向构成的表面比例成比例地增加。这允许纳米沉淀物采取更球形的形式,从而减少其总表面积。此外,所有铜纳米沉淀半径的 EAM 电位预测的 Fe-Cu 界面应变足够小,它们不会显着改变计算的界面能量密度值。










































 京公网安备 11010802027423号
京公网安备 11010802027423号