当前位置:
X-MOL 学术
›
CrystEngComm
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Influence of solvent on crystal nucleation of benzocaine
CrystEngComm ( IF 2.6 ) Pub Date : 2020-11-11 , DOI: 10.1039/d0ce01306d Dominic Cheuk 1, 2, 3, 4, 5 , Jacek Zeglinski 1, 2, 3, 4, 5 , Renuka Krishnaraj 1, 2, 3, 4, 5 , Åke C. Rasmuson 1, 2, 3, 4, 5
CrystEngComm ( IF 2.6 ) Pub Date : 2020-11-11 , DOI: 10.1039/d0ce01306d Dominic Cheuk 1, 2, 3, 4, 5 , Jacek Zeglinski 1, 2, 3, 4, 5 , Renuka Krishnaraj 1, 2, 3, 4, 5 , Åke C. Rasmuson 1, 2, 3, 4, 5
Affiliation
|
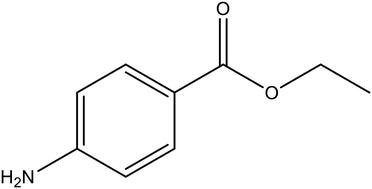
The influence of the solvent in nucleation of a polymorphic organic molecule, benzocaine, has been explored by measuring nucleation induction times, probing solvent–solute interactions in solution with spectroscopy and modelling the strength of solvent–solute intermolecular interactions using density functional theory (DFT). Over 2640 induction time experiments were conducted to study the crystal nucleation of benzocaine FII in six different organic solvents. The nucleation driving force required to reach the same induction time is strongly solvent-dependent, increasing in the order: ethyl acetate < 1,4-dioxane < acetonitrile < methanol < n-butanol. Nucleation in toluene is reasonably easy but the exact position varies with the induction time. The order between the solvents overall corresponds to the order of increasing interfacial energy as determined within the classical nucleation theory. The shift of the FTIR carbonyl frequency reflecting the strength in the solvent–solute interaction decreases in the same order as the interfacial energy of benzocaine FII increases. This shift is corroborated by DFT calculated energies of binding one solvent molecule to the carboxyl group of benzocaine. An even better correlation of the influence of the solvent on the nucleation is provided by DFT calculated energy of binding the complete first solvation shell to the benzocaine molecule. The different methods reveal a consistent picture and suggest that the stronger the solvent binds to the benzocaine molecule in solution, the slower the nucleation becomes.
中文翻译:

溶剂对苯佐卡因晶体成核的影响
通过测量成核诱导时间,用光谱法探测溶液中的溶剂-溶质相互作用并使用密度泛函理论(DFT)建模溶剂-溶质分子间相互作用的强度,探索了溶剂对多晶型有机分子苯并卡因成核的影响。 。进行了超过2640次的诱导时间实验,以研究六种不同有机溶剂中苯佐卡因FII的晶体成核。达到相同诱导时间所需的成核驱动力强烈依赖于溶剂,顺序依次为:乙酸乙酯<1,4-二恶烷<乙腈<甲醇< n-丁醇。甲苯中的成核相当容易,但确切位置随诱导时间而变化。溶剂之间的顺序总体上对应于经典成核理论中确定的界面能增加的顺序。FTIR羰基频率的变化反映了溶剂与溶质之间相互作用的强度,其降低程度与苯并卡因FII的界面能增加的顺序相同。通过DFT计算的将一个溶剂分子结合到苯佐卡因的羧基上的能量证实了这一转变。DFT计算出的将完整的第一个溶剂化壳层与苯并卡因分子结合的能量提供了溶剂对成核作用的更好关联。
更新日期:2020-11-19
中文翻译:
溶剂对苯佐卡因晶体成核的影响
通过测量成核诱导时间,用光谱法探测溶液中的溶剂-溶质相互作用并使用密度泛函理论(DFT)建模溶剂-溶质分子间相互作用的强度,探索了溶剂对多晶型有机分子苯并卡因成核的影响。 。进行了超过2640次的诱导时间实验,以研究六种不同有机溶剂中苯佐卡因FII的晶体成核。达到相同诱导时间所需的成核驱动力强烈依赖于溶剂,顺序依次为:乙酸乙酯<1,4-二恶烷<乙腈<甲醇< n-丁醇。甲苯中的成核相当容易,但确切位置随诱导时间而变化。溶剂之间的顺序总体上对应于经典成核理论中确定的界面能增加的顺序。FTIR羰基频率的变化反映了溶剂与溶质之间相互作用的强度,其降低程度与苯并卡因FII的界面能增加的顺序相同。通过DFT计算的将一个溶剂分子结合到苯佐卡因的羧基上的能量证实了这一转变。DFT计算出的将完整的第一个溶剂化壳层与苯并卡因分子结合的能量提供了溶剂对成核作用的更好关联。










































 京公网安备 11010802027423号
京公网安备 11010802027423号