当前位置:
X-MOL 学术
›
Chem. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Ligand design by targeting a binding site water
Chemical Science ( IF 7.6 ) Pub Date : 2020-11-19 , DOI: 10.1039/d0sc04938g Pierre Matricon 1 , R Rama Suresh 2 , Zhan-Guo Gao 2 , Nicolas Panel 1 , Kenneth A Jacobson 2 , Jens Carlsson 1
Chemical Science ( IF 7.6 ) Pub Date : 2020-11-19 , DOI: 10.1039/d0sc04938g Pierre Matricon 1 , R Rama Suresh 2 , Zhan-Guo Gao 2 , Nicolas Panel 1 , Kenneth A Jacobson 2 , Jens Carlsson 1
Affiliation
|
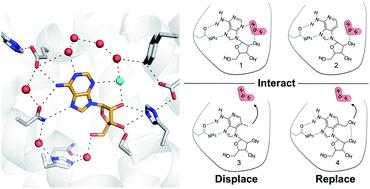
Solvent reorganization is a major driving force of protein–ligand association, but the contribution of binding site waters to ligand affinity is poorly understood. We investigated how altered interactions with a water network can influence ligand binding to a receptor. A series of ligands of the A2A adenosine receptor, which either interacted with or displaced an ordered binding site water, were studied experimentally and by molecular dynamics simulations. An analog of the endogenous ligand that was unable to hydrogen bond to the ordered water lost affinity and this activity cliff was captured by molecular dynamics simulations. Two compounds designed to displace the ordered water from the binding site were then synthesized and evaluated experimentally, leading to the discovery of an A2A agonist with nanomolar activity. Calculation of the thermodynamic profiles resulting from introducing substituents that interacted with or displaced the ordered water showed that the gain of binding affinity was enthalpy driven. Detailed analysis of the energetics and binding site hydration networks revealed that the enthalpy change was governed by contributions that are commonly neglected in structure-based drug optimization. In particular, simulations suggested that displacement of water from a binding site to the bulk solvent can lead to large energy contributions. Our findings provide insights into the molecular driving forces of protein–ligand binding and strategies for rational drug design.
中文翻译:

通过靶向结合位点水进行配体设计
溶剂重组是蛋白质-配体结合的主要驱动力,但结合位点水对配体亲和力的贡献知之甚少。我们研究了改变与水网络的相互作用如何影响配体与受体的结合。通过实验和分子动力学模拟研究了一系列 A 2A腺苷受体的配体,这些配体与有序的结合位点水相互作用或置换。不能与有序水形成氢键的内源性配体的类似物失去了亲和力,并且这种活性悬崖被分子动力学模拟捕获。然后合成了两种旨在从结合位点置换有序水的化合物并进行实验评估,从而发现了 A 2A具有纳摩尔活性的激动剂。由引入与有序水相互作用或取代有序水的取代基产生的热力学曲线的计算表明,结合亲和力的增益是焓驱动的。对能量学和结合位点水合网络的详细分析表明,焓变受基于结构的药物优化中通常被忽略的贡献支配。特别是,模拟表明水从结合位点置换到本体溶剂会导致较大的能量贡献。我们的研究结果提供了对蛋白质-配体结合的分子驱动力和合理药物设计策略的见解。
更新日期:2020-11-19
中文翻译:
通过靶向结合位点水进行配体设计
溶剂重组是蛋白质-配体结合的主要驱动力,但结合位点水对配体亲和力的贡献知之甚少。我们研究了改变与水网络的相互作用如何影响配体与受体的结合。通过实验和分子动力学模拟研究了一系列 A 2A腺苷受体的配体,这些配体与有序的结合位点水相互作用或置换。不能与有序水形成氢键的内源性配体的类似物失去了亲和力,并且这种活性悬崖被分子动力学模拟捕获。然后合成了两种旨在从结合位点置换有序水的化合物并进行实验评估,从而发现了 A 2A具有纳摩尔活性的激动剂。由引入与有序水相互作用或取代有序水的取代基产生的热力学曲线的计算表明,结合亲和力的增益是焓驱动的。对能量学和结合位点水合网络的详细分析表明,焓变受基于结构的药物优化中通常被忽略的贡献支配。特别是,模拟表明水从结合位点置换到本体溶剂会导致较大的能量贡献。我们的研究结果提供了对蛋白质-配体结合的分子驱动力和合理药物设计策略的见解。










































 京公网安备 11010802027423号
京公网安备 11010802027423号