Catalysis Communications ( IF 3.4 ) Pub Date : 2020-11-19 , DOI: 10.1016/j.catcom.2020.106245 Abdolhakim Pangh
|
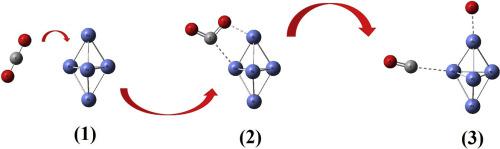
The chemisorption of CO2 molecule on metal clusters Ni4M (M = Ni, Mo, Sc, and Y) has been investigated by Density Functional Theory (DFT). The calculated adsorption energies were in the order of −73.3 kcal/mol and between −9.1 and −41 kcal/mol for pure Ni5 cluster and bimetallic clusters, respectively. Investigation of HOMO-LUMO energy gap in all complexes showed that the charge transfer occurs from the metallic clusters to the CO2 molecule. The rate constant kTST for the dissociation of CO2 molecule on the metal clusters under various pathways has been calculated. The Ni4Mo cluster acts as an inhibitor while the other metal clusters act as promoters.
中文翻译:
DFT研究:双金属Ni 4 M(M = Ni,Mo,Sc和Y)纳米簇上的CO 2催化裂解
通过密度泛函理论(DFT)研究了CO 2分子在金属簇Ni 4 M(M = Ni,Mo,Sc和Y)上的化学吸附。对于纯Ni 5团簇和双金属团簇,计算的吸附能分别为-73.3 kcal / mol的量级和-9.1至-41 kcal / mol的量级。对所有配合物中HOMO-LUMO能隙的研究表明,电荷从金属簇向CO 2分子转移。计算了在各种途径下金属簇上CO 2分子解离的速率常数k TST。镍4Mo团簇起抑制剂作用,而其他金属团簇起促进剂作用。










































 京公网安备 11010802027423号
京公网安备 11010802027423号