Chemical Physics Letters ( IF 2.8 ) Pub Date : 2020-11-18 , DOI: 10.1016/j.cplett.2020.138199 Tomoaki Kaneko , Keitaro Sodeyama
|
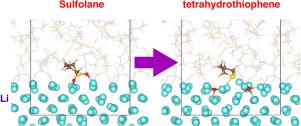
Sulfolane (SL) has attracted a great deal of interest as an electrolyte solvent in Li-metal batteries owing to its suitable properties. In this study, we performed a first-principles molecular dynamics simulation involving an Li metal LiB-SL liquid interface. It was found that two SO bonds of SL preferentially dissociate on the Li surface, while decomposition of B was not observed. Since the dissociated O atoms dissolved into the Li surface, our results indicate that an Li-oxide layer formed on the Li surface, which suggests that a preferable performance of the Li-metal negative electrode can be expected for this system.
中文翻译:
S的第一性原理分子动力学研究环丁砜在锂金属负极上的O键解离
环丁砜(SL)由于其合适的性能而在锂金属电池中作为电解质溶剂引起了广泛的关注。在这项研究中,我们进行了涉及锂金属的第一性原理分子动力学模拟 锂离子电池-SL液体接口。发现两个SSL的O键优先在Li表面解离,而B分解没有被观察到。由于解离的O原子溶解在Li表面中,因此我们的结果表明在Li表面上形成了一层Li-氧化物层,这表明可以为该系统预期Li-金属负极的性能。








































 京公网安备 11010802027423号
京公网安备 11010802027423号