当前位置:
X-MOL 学术
›
J. Phys. Chem. B
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Revealing the Inhibition Mechanism of RNA-Dependent RNA Polymerase (RdRp) of SARS-CoV-2 by Remdesivir and Nucleotide Analogues: A Molecular Dynamics Simulation Study
The Journal of Physical Chemistry B ( IF 3.3 ) Pub Date : 2020-11-15 , DOI: 10.1021/acs.jpcb.0c06747 Padmaja D. Wakchaure 1, 2 , Shibaji Ghosh 1, 2 , Bishwajit Ganguly 1, 2
The Journal of Physical Chemistry B ( IF 3.3 ) Pub Date : 2020-11-15 , DOI: 10.1021/acs.jpcb.0c06747 Padmaja D. Wakchaure 1, 2 , Shibaji Ghosh 1, 2 , Bishwajit Ganguly 1, 2
Affiliation
|
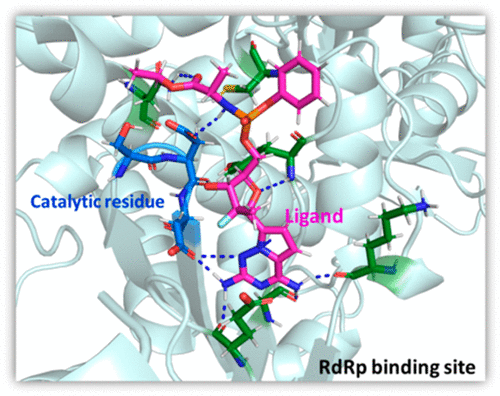
Antiviral drug therapy against SARS-CoV-2 is not yet established and posing a serious global health issue. Remdesivir is the first antiviral compound approved by the US FDA for the SARS-CoV-2 treatment for emergency use, targeting RNA-dependent RNA polymerase (RdRp) enzyme. In this work, we have examined the action of remdesivir and other two ligands screened from the library of nucleotide analogues using docking and molecular dynamics (MD) simulation studies. The MD simulations have been performed for all the ligand-bound RdRp complexes for the 30 ns time scale. This is one of the earlier reports to perform the MD simulations studies using the SARS-CoV-2 RdRp crystal structure (PDB ID 7BTF). The MD trajectories were analyzed and Molecular Mechanics Poisson–Boltzmann Surface Area (MM-PBSA) calculations were performed to calculate the binding free energy. The binding energy data reveal that compound-17 (−59.6 kcal/mol) binds more strongly as compared to compound-8 (−46.3 kcal/mol) and remdesivir (−29.7 kcal/mol) with RdRp. The detailed analysis of trajectories shows that the remdesivir binds in the catalytic site and forms a hydrogen bond with the catalytic residues from 0 to 0.46 ns. Compound-8 binds in the catalytic site but does not form direct hydrogen bonds with catalytic residues. Compound-17 showed the formation of hydrogen bonds with catalytic residues throughout the simulation process. The MD simulation results such as hydrogen bonding, the center of mass distance analysis, snapshots at a different time interval, and binding energy suggest that compound-17 binds strongly with RdRp of SARS-CoV-2 and has the potential to develop as a new antiviral against COVID-19. Further, the frontier molecular orbital analysis and molecular electrostatic potential (MESP) iso-surface analysis using DFT calculations shed light on the superior binding of compound-17 with RdRp compared to remdesivir and compound-8. The computed as well as the experimentally reported pharmacokinetics and toxicity parameters of compound-17 is encouraging and therefore can be one of the potential candidates for the treatment of COVID-19.
中文翻译:

伦德西韦和核苷酸类似物揭示SARS-CoV-2的RNA依赖性RNA聚合酶(RdRp)的抑制机制:分子动力学模拟研究。
尚未建立针对SARS-CoV-2的抗病毒药物治疗,并构成了严重的全球健康问题。Remdesivir是第一种被美国FDA批准用于SARS-CoV-2紧急治疗的抗病毒化合物,其靶向RNA依赖性RNA聚合酶(RdRp)酶。在这项工作中,我们使用对接和分子动力学(MD)模拟研究,研究了从核苷酸类似物库中筛选出的remdesivir和其他两个配体的作用。对所有配体结合的RdRp复合物进行了30 ns时间范围的MD模拟。这是使用SARS-CoV-2 RdRp晶体结构(PDB ID 7BTF)进行MD模拟研究的较早报告之一。分析了MD轨迹,并进行了分子力学泊松-玻尔兹曼表面积(MM-PBSA)计算以计算结合自由能。结合能数据显示,与具有RdRp的化合物8(-46.3 kcal / mol)和瑞姆昔韦(-29.7 kcal / mol)相比,化合物17(-59.6 kcal / mol)的结合更牢固。轨迹的详细分析表明,雷姆昔韦在催化位点结合并与催化残基形成0至0.46 ns的氢键。化合物-8在催化位点结合,但不与催化残基形成直接氢键。在整个模拟过程中,化合物17显示形成带有催化残基的氢键。MD模拟结果,例如氢键,质心距离分析,不同时间间隔的快照,和结合能表明,化合物17与SARS-CoV-2的RdRp牢固结合,并有可能发展为对抗COVID-19的新抗病毒药。此外,与伦贝昔韦和化合物8相比,使用DFT计算进行的前沿分子轨道分析和分子静电势(MESP)等值面分析揭示了化合物17与RdRp的优异结合。化合物17的计算以及实验报道的药代动力学和毒性参数令人鼓舞,因此可以成为治疗COVID-19的潜在候选药物之一。使用DFT计算进行的前沿分子轨道分析和分子静电势(MESP)等值面分析,揭示了化合物17与RdRp的结合优于伦德昔韦和化合物8。化合物17的计算以及实验报道的药代动力学和毒性参数令人鼓舞,因此可以成为治疗COVID-19的潜在候选药物之一。使用DFT计算进行的前沿分子轨道分析和分子静电势(MESP)等值面分析,揭示了化合物17与RdRp的结合优于伦德昔韦和化合物8。化合物17的计算以及实验报道的药代动力学和毒性参数令人鼓舞,因此可以成为治疗COVID-19的潜在候选药物之一。
更新日期:2020-11-25
中文翻译:
伦德西韦和核苷酸类似物揭示SARS-CoV-2的RNA依赖性RNA聚合酶(RdRp)的抑制机制:分子动力学模拟研究。
尚未建立针对SARS-CoV-2的抗病毒药物治疗,并构成了严重的全球健康问题。Remdesivir是第一种被美国FDA批准用于SARS-CoV-2紧急治疗的抗病毒化合物,其靶向RNA依赖性RNA聚合酶(RdRp)酶。在这项工作中,我们使用对接和分子动力学(MD)模拟研究,研究了从核苷酸类似物库中筛选出的remdesivir和其他两个配体的作用。对所有配体结合的RdRp复合物进行了30 ns时间范围的MD模拟。这是使用SARS-CoV-2 RdRp晶体结构(PDB ID 7BTF)进行MD模拟研究的较早报告之一。分析了MD轨迹,并进行了分子力学泊松-玻尔兹曼表面积(MM-PBSA)计算以计算结合自由能。结合能数据显示,与具有RdRp的化合物8(-46.3 kcal / mol)和瑞姆昔韦(-29.7 kcal / mol)相比,化合物17(-59.6 kcal / mol)的结合更牢固。轨迹的详细分析表明,雷姆昔韦在催化位点结合并与催化残基形成0至0.46 ns的氢键。化合物-8在催化位点结合,但不与催化残基形成直接氢键。在整个模拟过程中,化合物17显示形成带有催化残基的氢键。MD模拟结果,例如氢键,质心距离分析,不同时间间隔的快照,和结合能表明,化合物17与SARS-CoV-2的RdRp牢固结合,并有可能发展为对抗COVID-19的新抗病毒药。此外,与伦贝昔韦和化合物8相比,使用DFT计算进行的前沿分子轨道分析和分子静电势(MESP)等值面分析揭示了化合物17与RdRp的优异结合。化合物17的计算以及实验报道的药代动力学和毒性参数令人鼓舞,因此可以成为治疗COVID-19的潜在候选药物之一。使用DFT计算进行的前沿分子轨道分析和分子静电势(MESP)等值面分析,揭示了化合物17与RdRp的结合优于伦德昔韦和化合物8。化合物17的计算以及实验报道的药代动力学和毒性参数令人鼓舞,因此可以成为治疗COVID-19的潜在候选药物之一。使用DFT计算进行的前沿分子轨道分析和分子静电势(MESP)等值面分析,揭示了化合物17与RdRp的结合优于伦德昔韦和化合物8。化合物17的计算以及实验报道的药代动力学和毒性参数令人鼓舞,因此可以成为治疗COVID-19的潜在候选药物之一。


























 京公网安备 11010802027423号
京公网安备 11010802027423号