当前位置:
X-MOL 学术
›
J. Phys. Chem. B
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
NMR 1H–1H Dipole Relaxation in Fluids: Relaxation of Individual 1H–1H Pairs versus Relaxation of Molecular Modes
The Journal of Physical Chemistry B ( IF 2.8 ) Pub Date : 2020-11-13 , DOI: 10.1021/acs.jpcb.0c08078 D. Asthagiri 1 , Walter G. Chapman 1 , George J. Hirasaki 1 , Philip M. Singer 1
The Journal of Physical Chemistry B ( IF 2.8 ) Pub Date : 2020-11-13 , DOI: 10.1021/acs.jpcb.0c08078 D. Asthagiri 1 , Walter G. Chapman 1 , George J. Hirasaki 1 , Philip M. Singer 1
Affiliation
|
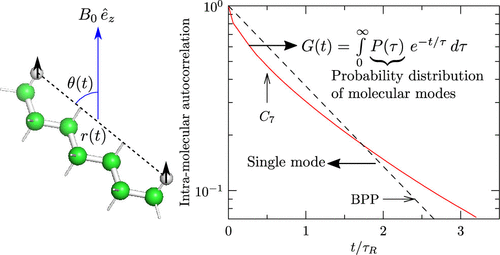
The intramolecular 1H NMR dipole–dipole relaxation of molecular fluids has traditionally been interpreted within the Bloembergen–Purcell–Pound (BPP) theory of NMR intramolecular relaxation. The BPP theory draws upon Debye’s theory for describing the rotational diffusion of the 1H–1H pair and predicts a monoexponential decay of the 1H–1H dipole–dipole autocorrelation function between distinct spin pairs. Using molecular dynamics (MD) simulations, we show that for both n-heptane and water this is not the case. In particular, the autocorrelation function of individual 1H–1H intramolecular pairs itself evinces a rich stretched-exponential behavior, implying a distribution in rotational correlation times. However, for the high-symmetry molecule neopentane, the individual 1H–1H intramolecular pairs do conform to the BPP description, suggesting an important role of molecular symmetry in aiding agreement with the BPP model. The intermolecular autocorrelation functions for n-heptane, water, and neopentane also do not admit a monoexponential behavior of individual 1H–1H intermolecular pairs at distinct initial separations. We suggest expanding the autocorrelation function in terms of modes, provisionally termed molecular modes, that do have an exponential relaxation behavior. With care, the resulting Fredholm integral equation of the first kind can be inverted to recover the probability distribution of the molecular modes. The advantages and limitations of this approach are noted.
中文翻译:

流体中的NMR 1 H– 1 H偶极弛豫:单个1 H– 1 H对的弛豫与分子模式的弛豫
传统上,分子流体的分子内1 H NMR偶极-偶极弛豫在NMR分子内弛豫的Bloembergen-Purcell-Pound(BPP)理论中得到了解释。的BPP理论借鉴德拜的理论用于说明的旋转扩散1 H- 1 ħ对和预测的单指数衰减1和H- 1不同自旋对H之间的偶极-偶极自相关函数。使用分子动力学(MD)模拟,我们发现对于正庚烷和水而言,情况并非如此。特别是个体1 H– 1的自相关函数H个分子内对本身表明有丰富的拉伸指数行为,这意味着旋转相关时间的分布。但是,对于高对称分子新戊烷,单个的1 H– 1 H分子内对确实符合BPP的描述,这表明分子对称在帮助与BPP模型达成一致方面具有重要作用。正庚烷,水和新戊烷的分子间自相关函数也不允许单个1 H– 1的单指数行为H个分子间对处于不同的初始间隔。我们建议根据确实具有指数弛豫行为的模式(临时称为分子模式)扩展自相关函数。小心地,可以对所得的第一类Fredholm积分方程进行求逆,以恢复分子模态的概率分布。指出了此方法的优点和局限性。
更新日期:2020-11-25
中文翻译:
流体中的NMR 1 H– 1 H偶极弛豫:单个1 H– 1 H对的弛豫与分子模式的弛豫
传统上,分子流体的分子内1 H NMR偶极-偶极弛豫在NMR分子内弛豫的Bloembergen-Purcell-Pound(BPP)理论中得到了解释。的BPP理论借鉴德拜的理论用于说明的旋转扩散1 H- 1 ħ对和预测的单指数衰减1和H- 1不同自旋对H之间的偶极-偶极自相关函数。使用分子动力学(MD)模拟,我们发现对于正庚烷和水而言,情况并非如此。特别是个体1 H– 1的自相关函数H个分子内对本身表明有丰富的拉伸指数行为,这意味着旋转相关时间的分布。但是,对于高对称分子新戊烷,单个的1 H– 1 H分子内对确实符合BPP的描述,这表明分子对称在帮助与BPP模型达成一致方面具有重要作用。正庚烷,水和新戊烷的分子间自相关函数也不允许单个1 H– 1的单指数行为H个分子间对处于不同的初始间隔。我们建议根据确实具有指数弛豫行为的模式(临时称为分子模式)扩展自相关函数。小心地,可以对所得的第一类Fredholm积分方程进行求逆,以恢复分子模态的概率分布。指出了此方法的优点和局限性。










































 京公网安备 11010802027423号
京公网安备 11010802027423号