当前位置:
X-MOL 学术
›
Comp. Mater. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Monte Carlo simulation of order-disorder transition in refractory high entropy alloys: A data-driven approach
Computational Materials Science ( IF 3.1 ) Pub Date : 2021-02-01 , DOI: 10.1016/j.commatsci.2020.110135 Xianglin Liu , Jiaxin Zhang , Junqi Yin , Sirui Bi , Markus Eisenbach , Yang Wang
Computational Materials Science ( IF 3.1 ) Pub Date : 2021-02-01 , DOI: 10.1016/j.commatsci.2020.110135 Xianglin Liu , Jiaxin Zhang , Junqi Yin , Sirui Bi , Markus Eisenbach , Yang Wang
|
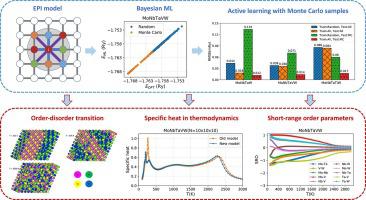
Abstract High entropy alloys (HEAs) are a series of novel materials that demonstrate many exceptional mechanical properties. To understand the origin of these attractive properties, it is important to investigate the thermodynamics and elucidate the evolution of various chemical phases. In this work, we introduce a data-driven approach to construct the effective Hamiltonian and study the thermodynamics of HEAs through canonical Monte Carlo simulation. The main characteristic of our method is to use pairwise interactions between atoms as features and systematically improve the representativeness of the dataset using samples from Monte Carlo simulation. We find this method produces highly robust and accurate effective Hamiltonians that give less than 0.1 mRy test error for all the three refractory HEAs: MoNbTaW, MoNbTaVW, and MoNbTaTiW. Using replica exchange to speed up the MC simulation, we calculated the specific heats and short-range order parameters in a wide range of temperatures. For all the studied materials, we find there are two major order-disorder transitions occurring respectively at T 1 and T 2 , where T 1 is near room temperature but T 2 is much higher. We further demonstrate that the transition at T 1 is caused by W and Nb while the one at T 2 is caused by the other elements. By comparing with experiments, the results provide insight into the role of chemical ordering in the strength and ductility of HEAs.
中文翻译:

难熔高熵合金有序-无序转变的蒙特卡罗模拟:数据驱动的方法
摘要 高熵合金 (HEAs) 是一系列具有许多优异机械性能的新型材料。要了解这些吸引人的特性的起源,重要的是研究热力学并阐明各种化学相的演变。在这项工作中,我们引入了一种数据驱动的方法来构建有效的哈密顿量,并通过规范的蒙特卡罗模拟研究 HEA 的热力学。我们方法的主要特点是使用原子之间的成对相互作用作为特征,并使用蒙特卡罗模拟的样本系统地提高数据集的代表性。我们发现这种方法产生了高度稳健和准确的有效哈密顿量,对于所有三种难熔 HEA:MoNbTaW、MoNbTaVW 和 MoNbTaTiW,测试误差小于 0.1 mRy。使用副本交换来加速 MC 模拟,我们计算了较宽温度范围内的比热和短程有序参数。对于所有研究的材料,我们发现分别在 T 1 和 T 2 处发生了两个主要的有序-无序转变,其中 T 1 接近室温但 T 2 高得多。我们进一步证明 T 1 处的跃迁是由 W 和 Nb 引起的,而 T 2 处的跃迁是由其他元素引起的。通过与实验的比较,结果提供了对化学排序在 HEA 强度和延展性中的作用的深入了解。我们发现分别在 T 1 和 T 2 处发生了两个主要的有序-无序转变,其中 T 1 接近室温但 T 2 高得多。我们进一步证明 T 1 处的跃迁是由 W 和 Nb 引起的,而 T 2 处的跃迁是由其他元素引起的。通过与实验的比较,结果提供了对化学排序在 HEA 强度和延展性中的作用的深入了解。我们发现分别在 T 1 和 T 2 处发生了两个主要的有序-无序转变,其中 T 1 接近室温但 T 2 高得多。我们进一步证明 T 1 处的跃迁是由 W 和 Nb 引起的,而 T 2 处的跃迁是由其他元素引起的。通过与实验的比较,结果提供了对化学排序在 HEA 强度和延展性中的作用的深入了解。
更新日期:2021-02-01
中文翻译:
难熔高熵合金有序-无序转变的蒙特卡罗模拟:数据驱动的方法
摘要 高熵合金 (HEAs) 是一系列具有许多优异机械性能的新型材料。要了解这些吸引人的特性的起源,重要的是研究热力学并阐明各种化学相的演变。在这项工作中,我们引入了一种数据驱动的方法来构建有效的哈密顿量,并通过规范的蒙特卡罗模拟研究 HEA 的热力学。我们方法的主要特点是使用原子之间的成对相互作用作为特征,并使用蒙特卡罗模拟的样本系统地提高数据集的代表性。我们发现这种方法产生了高度稳健和准确的有效哈密顿量,对于所有三种难熔 HEA:MoNbTaW、MoNbTaVW 和 MoNbTaTiW,测试误差小于 0.1 mRy。使用副本交换来加速 MC 模拟,我们计算了较宽温度范围内的比热和短程有序参数。对于所有研究的材料,我们发现分别在 T 1 和 T 2 处发生了两个主要的有序-无序转变,其中 T 1 接近室温但 T 2 高得多。我们进一步证明 T 1 处的跃迁是由 W 和 Nb 引起的,而 T 2 处的跃迁是由其他元素引起的。通过与实验的比较,结果提供了对化学排序在 HEA 强度和延展性中的作用的深入了解。我们发现分别在 T 1 和 T 2 处发生了两个主要的有序-无序转变,其中 T 1 接近室温但 T 2 高得多。我们进一步证明 T 1 处的跃迁是由 W 和 Nb 引起的,而 T 2 处的跃迁是由其他元素引起的。通过与实验的比较,结果提供了对化学排序在 HEA 强度和延展性中的作用的深入了解。我们发现分别在 T 1 和 T 2 处发生了两个主要的有序-无序转变,其中 T 1 接近室温但 T 2 高得多。我们进一步证明 T 1 处的跃迁是由 W 和 Nb 引起的,而 T 2 处的跃迁是由其他元素引起的。通过与实验的比较,结果提供了对化学排序在 HEA 强度和延展性中的作用的深入了解。









































 京公网安备 11010802027423号
京公网安备 11010802027423号