当前位置:
X-MOL 学术
›
Comp. Mater. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Fitting of interatomic potentials by a differential evolution algorithm
Computational Materials Science ( IF 3.1 ) Pub Date : 2021-02-01 , DOI: 10.1016/j.commatsci.2020.109929 Giovani L. Rech , André L. Martinotto , Naira M. Balzaretti , Cláudio A. Perottoni
Computational Materials Science ( IF 3.1 ) Pub Date : 2021-02-01 , DOI: 10.1016/j.commatsci.2020.109929 Giovani L. Rech , André L. Martinotto , Naira M. Balzaretti , Cláudio A. Perottoni
|
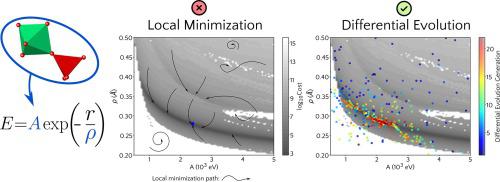
Abstract Computer simulation has been increasingly present in the discovery and understanding of materials structure and properties. First-principles simulations, in particular, allow to meaningfully explore the material world without having to resource to experimental methods. Even though their extreme usefulness, first-principle methods tend to be very computer-intensive to the point of being impractical in many situations that are important for the understanding of materials’ behavior. As an alternative, computer simulations can be carried out using interatomic potentials (IPs), in which case an analytic mathematical model for the energy of the system is fitted to a set of crystal structure parameters and experimentally determined physical properties by minimizing a cost function. How close the properties calculated using an IP can get of those experimentally determined is limited both by its analytic form and the model’s parameters. For compounds with different atomic species, the multidimensional parameter space of the cost function most possibly exhibit a very complex landscape. In these cases, global minimization methods are often required. This work explores the use of a differential evolution (DE) algorithm for the parametrization of IPs for two compounds, namely berlinite (AlPO4) and zirconium tungstate ( α -ZrW2O8). Several analytic interatomic potentials (including some previously proposed in the literature) were fitted to experimental lattice parameters, atomic positions, and elastic constants. Two-dimensional mappings of the interatomic potential parameters reveal the complex landscape of the cost function for berlinite and α -ZrW8O8, which exhibits several local minima, broad plateaus, discontinuities, and, in some cases, strong correlations between fitting parameters. The differential evolution algorithm was able to navigate the cost function landscape and, giving a sufficiently large population, was capable to found good candidates for the global minimum, despite all the mentioned difficulties. In all cases here explored, the interatomic potentials re-parametrized using DE yield calculated athermal properties in better agreement with the experimental observables as compared to previous results from the literature.
中文翻译:

通过微分进化算法拟合原子间势
摘要 计算机模拟越来越多地出现在材料结构和性能的发现和理解中。尤其是第一性原理模拟,可以有意义地探索物质世界,而无需借助实验方法。尽管它们极其有用,但第一性原理方法往往是计算机密集型的,以至于在许多对理解材料行为很重要的情况下是不切实际的。作为替代方案,可以使用原子间势 (IP) 进行计算机模拟,在这种情况下,系统能量的解析数学模型适合一组晶体结构参数和通过最小化成本函数通过实验确定的物理特性。使用 IP 计算的属性与实验确定的属性的接近程度受其分析形式和模型参数的限制。对于具有不同原子种类的化合物,成本函数的多维参数空间很可能表现出非常复杂的景观。在这些情况下,通常需要全局最小化方法。这项工作探索了使用差分进化 (DE) 算法对两种化合物的 IP 进行参数化,即伯林石 (AlPO4) 和钨酸锆 (α -ZrW2O8)。几个分析原子间势(包括一些先前在文献中提出的)被拟合到实验晶格参数、原子位置和弹性常数。原子间势参数的二维映射揭示了 berlinite 和 α -ZrW8O8 成本函数的复杂景观,它表现出几个局部最小值、宽阔的高原、不连续性,以及在某些情况下,拟合参数之间的强相关性。尽管存在所有提到的困难,但差分进化算法能够导航成本函数景观,并且在提供足够大的种群的情况下,能够为全局最小值找到好的候选者。在此处探讨的所有情况下,与文献中的先前结果相比,使用 DE 产率计算的非热特性重新参数化的原子间势与实验观测值更好地吻合。拟合参数之间的强相关性。尽管存在所有提到的困难,但差分进化算法能够导航成本函数景观,并且在提供足够大的种群的情况下,能够为全局最小值找到好的候选者。在此处探讨的所有情况下,与文献中的先前结果相比,使用 DE 产率计算的非热特性重新参数化的原子间势与实验观测值更好地吻合。拟合参数之间的强相关性。尽管存在所有提到的困难,但差分进化算法能够导航成本函数景观,并且在提供足够大的种群的情况下,能够为全局最小值找到好的候选者。在此处探讨的所有情况下,与文献中的先前结果相比,使用 DE 产率计算的非热特性重新参数化的原子间势与实验观测值更好地吻合。
更新日期:2021-02-01
中文翻译:
通过微分进化算法拟合原子间势
摘要 计算机模拟越来越多地出现在材料结构和性能的发现和理解中。尤其是第一性原理模拟,可以有意义地探索物质世界,而无需借助实验方法。尽管它们极其有用,但第一性原理方法往往是计算机密集型的,以至于在许多对理解材料行为很重要的情况下是不切实际的。作为替代方案,可以使用原子间势 (IP) 进行计算机模拟,在这种情况下,系统能量的解析数学模型适合一组晶体结构参数和通过最小化成本函数通过实验确定的物理特性。使用 IP 计算的属性与实验确定的属性的接近程度受其分析形式和模型参数的限制。对于具有不同原子种类的化合物,成本函数的多维参数空间很可能表现出非常复杂的景观。在这些情况下,通常需要全局最小化方法。这项工作探索了使用差分进化 (DE) 算法对两种化合物的 IP 进行参数化,即伯林石 (AlPO4) 和钨酸锆 (α -ZrW2O8)。几个分析原子间势(包括一些先前在文献中提出的)被拟合到实验晶格参数、原子位置和弹性常数。原子间势参数的二维映射揭示了 berlinite 和 α -ZrW8O8 成本函数的复杂景观,它表现出几个局部最小值、宽阔的高原、不连续性,以及在某些情况下,拟合参数之间的强相关性。尽管存在所有提到的困难,但差分进化算法能够导航成本函数景观,并且在提供足够大的种群的情况下,能够为全局最小值找到好的候选者。在此处探讨的所有情况下,与文献中的先前结果相比,使用 DE 产率计算的非热特性重新参数化的原子间势与实验观测值更好地吻合。拟合参数之间的强相关性。尽管存在所有提到的困难,但差分进化算法能够导航成本函数景观,并且在提供足够大的种群的情况下,能够为全局最小值找到好的候选者。在此处探讨的所有情况下,与文献中的先前结果相比,使用 DE 产率计算的非热特性重新参数化的原子间势与实验观测值更好地吻合。拟合参数之间的强相关性。尽管存在所有提到的困难,但差分进化算法能够导航成本函数景观,并且在提供足够大的种群的情况下,能够为全局最小值找到好的候选者。在此处探讨的所有情况下,与文献中的先前结果相比,使用 DE 产率计算的非热特性重新参数化的原子间势与实验观测值更好地吻合。










































 京公网安备 11010802027423号
京公网安备 11010802027423号