Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Multiple Molecular Dynamics Simulations of the Inhibitor GRL-02031 Complex with Wild Type and Mutant HIV-1 Protease Reveal the Binding and Drug-Resistance Mechanism
Langmuir ( IF 3.7 ) Pub Date : 2020-11-11 , DOI: 10.1021/acs.langmuir.0c02151 Ruige Wang 1 , Qingchuan Zheng 1, 2
Langmuir ( IF 3.7 ) Pub Date : 2020-11-11 , DOI: 10.1021/acs.langmuir.0c02151 Ruige Wang 1 , Qingchuan Zheng 1, 2
Affiliation
|
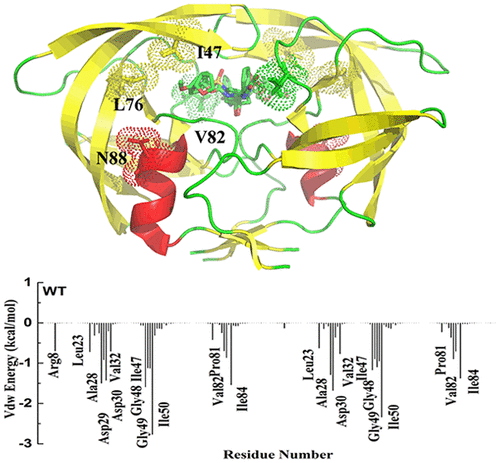
Human immunodeficiency virus type 1 (HIV-1) protease is regarded as a fascinating target for drug development against HIV infection. However, mutations causing drug resistance severely limit the efficiency of the recently marketed drugs in the treatment of HIV replication. To elucidate the binding mechanism of HIV-1 protease with promising inhibitor GRL-02031 and further to probe the resistance mechanism associated with mutations (I47V, L76V, V82A, and N88D) to the inhibitor, we applied multiple molecular dynamics (MMD) simulations along with energy analysis by the molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) and solvated interaction energy (SIE) methodology on specific HIV-1 protease with GRL-0231 complexes. On the basis of detail analysis of the simulations, we revealed key characteristics that constitute the drug resistance of four mutation HIV-1 proteases toward GRL-02031: substitution of the side chain in these four mutation residues leads to a change in the distances between the flaps and catalytic sites, thereby reducing the affinity for GRL-02031 with these four mutation proteases, even though the L76V and N88D residues cannot directly contact GRL-02031. The results of energy analysis according to the MM-PBSA and SIE methods further indicated that hydrophobic interaction was considered to be the prime driving force for inhibitor GRL-02031 binding to protease and the decrease in van der Waals interactions between inhibitor GRL-02031 and mutant proteases as the primary cause of the drug resistance. Analyses of the hydrogen bonds and atomic interactions further provided detailed explanations for the resistance of these four mutation proteases toward inhibitor GRL-02031. The present study provides potential guidance on the structure-based inhibitors’ design targeting HIV-1 protease.
中文翻译:

具有野生型和突变HIV-1蛋白酶的抑制剂GRL-02031配合物的多重分子动力学模拟揭示了结合和耐药机制
人类免疫缺陷病毒1型(HIV-1)蛋白酶被认为是抗HIV感染药物开发的迷人目标。但是,引起耐药性的突变严重限制了最近上市的药物在治疗HIV复制中的效率。为了阐明HIV-1蛋白酶与有希望的抑制剂GRL-02031的结合机制,并进一步探索与突变(I47V,L76V,V82A和N88D)对该抑制剂产生的抗药性机制,我们沿用了多种分子动力学(MMD)模拟通过分子力学泊松-玻尔兹曼表面积(MM-PBSA)和溶剂化的相互作用能(SIE)方法对具有GRL-0231复合物的特定HIV-1蛋白酶进行了能量分析。在对仿真进行详细分析的基础上,我们揭示了构成四个突变HIV-1蛋白酶对GRL-02031的耐药性的关键特征:这四个突变残基中的侧链取代导致襟翼和催化位点之间的距离发生变化,从而降低了对GRL-02031具有这四种突变蛋白酶,即使L76V和N88D残基不能直接接触GRL-02031。根据MM-PBSA和SIE方法进行的能量分析结果进一步表明,疏水相互作用被认为是抑制剂GRL-02031与蛋白酶结合的主要驱动力,并且抑制剂GRL-02031与突变体之间的范德华相互作用降低蛋白酶是引起耐药性的主要原因。氢键和原子相互作用的分析进一步提供了这四种突变蛋白酶对抑制剂GRL-02031的抗性的详细解释。本研究为针对HIV-1蛋白酶的基于结构的抑制剂的设计提供了潜在的指导。
更新日期:2020-11-25
中文翻译:
具有野生型和突变HIV-1蛋白酶的抑制剂GRL-02031配合物的多重分子动力学模拟揭示了结合和耐药机制
人类免疫缺陷病毒1型(HIV-1)蛋白酶被认为是抗HIV感染药物开发的迷人目标。但是,引起耐药性的突变严重限制了最近上市的药物在治疗HIV复制中的效率。为了阐明HIV-1蛋白酶与有希望的抑制剂GRL-02031的结合机制,并进一步探索与突变(I47V,L76V,V82A和N88D)对该抑制剂产生的抗药性机制,我们沿用了多种分子动力学(MMD)模拟通过分子力学泊松-玻尔兹曼表面积(MM-PBSA)和溶剂化的相互作用能(SIE)方法对具有GRL-0231复合物的特定HIV-1蛋白酶进行了能量分析。在对仿真进行详细分析的基础上,我们揭示了构成四个突变HIV-1蛋白酶对GRL-02031的耐药性的关键特征:这四个突变残基中的侧链取代导致襟翼和催化位点之间的距离发生变化,从而降低了对GRL-02031具有这四种突变蛋白酶,即使L76V和N88D残基不能直接接触GRL-02031。根据MM-PBSA和SIE方法进行的能量分析结果进一步表明,疏水相互作用被认为是抑制剂GRL-02031与蛋白酶结合的主要驱动力,并且抑制剂GRL-02031与突变体之间的范德华相互作用降低蛋白酶是引起耐药性的主要原因。氢键和原子相互作用的分析进一步提供了这四种突变蛋白酶对抑制剂GRL-02031的抗性的详细解释。本研究为针对HIV-1蛋白酶的基于结构的抑制剂的设计提供了潜在的指导。










































 京公网安备 11010802027423号
京公网安备 11010802027423号