当前位置:
X-MOL 学术
›
Cryst. Growth Des.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Understanding Reactivity and Assembly of Dichalcogenides: Structural, Electrostatic Potential, and Topological Analyses of 3H-1,2-Benzodithiol-3-one and Selenium Analogs
Crystal Growth & Design ( IF 3.2 ) Pub Date : 2020-11-09 , DOI: 10.1021/acs.cgd.0c00961 Rahul Shukla 1 , Arun Dhaka 2 , Emmanuel Aubert 1 , Vishnu Vijayakumar-Syamala 1 , Olivier Jeannin 2 , Marc Fourmigué 2 , Enrique Espinosa 1
Crystal Growth & Design ( IF 3.2 ) Pub Date : 2020-11-09 , DOI: 10.1021/acs.cgd.0c00961 Rahul Shukla 1 , Arun Dhaka 2 , Emmanuel Aubert 1 , Vishnu Vijayakumar-Syamala 1 , Olivier Jeannin 2 , Marc Fourmigué 2 , Enrique Espinosa 1
Affiliation
|
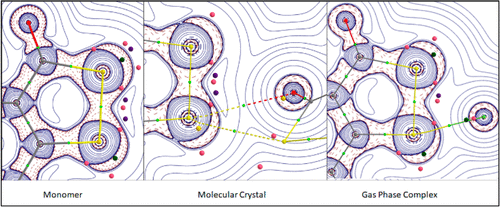
Molecular assembly and reactivity have been investigated with a series of 3H-1,2-benzodithiol-3-(thi)one derivatives and their (mixed) selenated analogs. Electrostatic potential calculations on monomers show three σ-hole regions around the dichalcogenide Ch–Ch bond (Ch = S, Se), one side-on and two along the bonding direction. The topological analysis of the electron density ρ(r) points to the weak nature of the Ch–Ch bond. σ-Hole and lone-pair regions are described in terms of charge depletion (CD) and charge concentration (CC) sites found in the valence shell of chalcogen atoms. Whereas CD and CC sites are characterized by the topological critical points of L(r) = −∇2ρ(r), their electrophilic and nucleophilic powers are measured by the corresponding L/ρ magnitudes. In crystal structures, each chalcogen bond (ChB) involves a σ-hole region and shows a CD···CC interaction that aligns with the internuclear direction of the atoms the CD and CC sites belong. The alignment holds simultaneously for all of the ChB interactions in each crystal structure, indicating that CD···CC interactions drive molecular orientation in molecular assembly. Strength of ChB is measured in terms of the topological properties of ρ(r), whereas the intensity of the electrophilic···nucleophilic interaction is monitored by [(L/ρ)CC – (L/ρ)CD]/dCC···CD2. The σ-hole in side-on conformation forms the strongest ChB interactions in molecular assembly. Reactivity of molecules against nucleophilic attack has been investigated along each of the three σ-hole regions by using fluoride as a probe. Adducts formed along the Ch–Ch bonding direction are energetically more favorable than in side-on conformation. At optimized geometries, the F···Ch bond (Ch = S, Se) exhibits a partial covalent character, while it weakens concomitantly the Ch···Ch bond that also becomes of partial covalent character. In the reactivity process, the significant reorientation of the plane containing the chalcogen lone pairs, along with the opening, shrinking, and splitting of reactivity surfaces ∇2ρ(r) = 0, is the signature of the charge redistribution that involves the nucleophilic attack.
中文翻译:

了解二硫属元素化物的反应性和组装:3 H -1,2-苯二硫基-3-酮和硒类似物的结构,静电势和拓扑分析
用一系列的3 H -1,2-苯并二硫基-3-(thi)one衍生物及其(混合的)硒化类似物研究了分子组装和反应性。单体的静电势计算结果表明,二卤化氢Ch-Ch键(Ch = S,Se)周围有三个σ孔区域,一侧朝上,两个沿结合方向。电子密度ρ(r)的拓扑分析指出了Ch–Ch键的弱性质。σ孔和孤对区是根据硫族元素原子的价壳中的电荷耗尽(CD)和电荷浓度(CC)位点描述的。而CD和CC位点的特征在于拓扑临界点大号([R)=-∇ 2 ρ(ř),它们的亲电和亲核能力通过相应的L /ρ值来衡量。在晶体结构中,每个硫族元素键(ChB)都包含一个σ孔区域,并显示出CD···CC相互作用,该相互作用与CD和CC位置所属的原子的核间方向对齐。对于每个晶体结构中的所有ChB相互作用,排列均同时保持,表明CD···CC相互作用驱动分子组装中的分子取向。ChB的强度是根据ρ(r)的拓扑性质来衡量的,而亲电···亲核相互作用的强度则通过[(L /ρ)CC –(L /ρ)CD ] / d CC·进行监测。··光盘2。侧面构象中的σ孔在分子组装中形成最强的ChB相互作用。通过使用氟化物作为探针,已经沿着三个σ孔区域的每一个研究了分子对亲核攻击的反应性。沿Ch-Ch键合方向形成的加合物在能量上比侧面构象更有利。在优化的几何构型下,F··Ch键(Ch = S,Se)表现出部分共价特征,同时削弱了也成为部分共价特征的Ch··Ch键。在反应过程中,平面的重取向显著含有硫属孤对,沿与开口,萎缩,和反应性的分裂表面∇ 2 ρ(ř)= 0,是涉及亲核攻击的电荷重新分布的特征。
更新日期:2020-12-02
中文翻译:
了解二硫属元素化物的反应性和组装:3 H -1,2-苯二硫基-3-酮和硒类似物的结构,静电势和拓扑分析
用一系列的3 H -1,2-苯并二硫基-3-(thi)one衍生物及其(混合的)硒化类似物研究了分子组装和反应性。单体的静电势计算结果表明,二卤化氢Ch-Ch键(Ch = S,Se)周围有三个σ孔区域,一侧朝上,两个沿结合方向。电子密度ρ(r)的拓扑分析指出了Ch–Ch键的弱性质。σ孔和孤对区是根据硫族元素原子的价壳中的电荷耗尽(CD)和电荷浓度(CC)位点描述的。而CD和CC位点的特征在于拓扑临界点大号([R)=-∇ 2 ρ(ř),它们的亲电和亲核能力通过相应的L /ρ值来衡量。在晶体结构中,每个硫族元素键(ChB)都包含一个σ孔区域,并显示出CD···CC相互作用,该相互作用与CD和CC位置所属的原子的核间方向对齐。对于每个晶体结构中的所有ChB相互作用,排列均同时保持,表明CD···CC相互作用驱动分子组装中的分子取向。ChB的强度是根据ρ(r)的拓扑性质来衡量的,而亲电···亲核相互作用的强度则通过[(L /ρ)CC –(L /ρ)CD ] / d CC·进行监测。··光盘2。侧面构象中的σ孔在分子组装中形成最强的ChB相互作用。通过使用氟化物作为探针,已经沿着三个σ孔区域的每一个研究了分子对亲核攻击的反应性。沿Ch-Ch键合方向形成的加合物在能量上比侧面构象更有利。在优化的几何构型下,F··Ch键(Ch = S,Se)表现出部分共价特征,同时削弱了也成为部分共价特征的Ch··Ch键。在反应过程中,平面的重取向显著含有硫属孤对,沿与开口,萎缩,和反应性的分裂表面∇ 2 ρ(ř)= 0,是涉及亲核攻击的电荷重新分布的特征。










































 京公网安备 11010802027423号
京公网安备 11010802027423号