当前位置:
X-MOL 学术
›
Comp. Mater. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Alternative algorithms for simultaneous modeling of ordering and intermediate compound growth during reactive diffusion
Computational Materials Science ( IF 3.1 ) Pub Date : 2021-02-01 , DOI: 10.1016/j.commatsci.2020.110114 Viktoriia Pasichna , Andriy Gusak
Computational Materials Science ( IF 3.1 ) Pub Date : 2021-02-01 , DOI: 10.1016/j.commatsci.2020.110114 Viktoriia Pasichna , Andriy Gusak
|
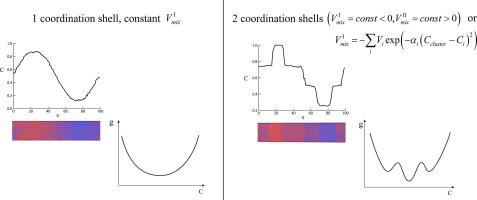
Abstract Standard Monte Carlo algorithms with constant pair interactions within the first coordination shell are usually quite effective for the description of ordering, tracer diffusion, chemical diffusion (but without Kirkendall shift effect) within any already existing ordered intermediate phase. Yet, one encounters big problems if one tries to simulate the formation and growth of ordered intermediate phase or, especially, simultaneous formation and growth of several intermediate phases during interdiffusion. Two algorithms providing solution of this problem, are discussed. The first algorithm is known (but not widely used for reactive growth) – account of interactions in the two coordination shells with negative mixing energy in the first coordination shell and positive mixing energy in the second one. The second algorithm is new: interactions only within the first coordination shell of each atom but depending on the local composition within the cluster around interacting atoms. Both algorithms are shown to provide formation and growth of the ordered intermediate phases with rather narrow concentration ranges, growing with time (after some initial period) according to parabolic law.
中文翻译:

反应扩散过程中有序和中间化合物生长同时建模的替代算法
摘要 在第一配位壳内具有恒定对相互作用的标准蒙特卡罗算法对于描述任何现有有序中间相内的排序、示踪剂扩散、化学扩散(但没有柯肯德尔位移效应)通常非常有效。然而,如果试图模拟有序中间相的形成和生长,或者特别是在相互扩散过程中同时形成和生长几个中间相,就会遇到大问题。讨论了解决这个问题的两种算法。第一种算法是已知的(但并未广泛用于反应性生长)——考虑两个配位壳中的相互作用,第一个配位壳中的混合能量为负,第二个配位壳中的混合能量为正。第二个算法是新的:相互作用仅在每个原子的第一配位壳内,但取决于相互作用原子周围簇内的局部组成。根据抛物线定律,两种算法都显示出具有相当窄的浓度范围的有序中间相的形成和生长,随时间增长(在某个初始阶段之后)。
更新日期:2021-02-01
中文翻译:
反应扩散过程中有序和中间化合物生长同时建模的替代算法
摘要 在第一配位壳内具有恒定对相互作用的标准蒙特卡罗算法对于描述任何现有有序中间相内的排序、示踪剂扩散、化学扩散(但没有柯肯德尔位移效应)通常非常有效。然而,如果试图模拟有序中间相的形成和生长,或者特别是在相互扩散过程中同时形成和生长几个中间相,就会遇到大问题。讨论了解决这个问题的两种算法。第一种算法是已知的(但并未广泛用于反应性生长)——考虑两个配位壳中的相互作用,第一个配位壳中的混合能量为负,第二个配位壳中的混合能量为正。第二个算法是新的:相互作用仅在每个原子的第一配位壳内,但取决于相互作用原子周围簇内的局部组成。根据抛物线定律,两种算法都显示出具有相当窄的浓度范围的有序中间相的形成和生长,随时间增长(在某个初始阶段之后)。










































 京公网安备 11010802027423号
京公网安备 11010802027423号