当前位置:
X-MOL 学术
›
Int. J. Quantum Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Insight into the X‐ray absorption spectra of Cu‐porphyrazines from electronic structure theory
International Journal of Quantum Chemistry ( IF 2.3 ) Pub Date : 2020-10-26 , DOI: 10.1002/qua.26515 Esma Birsen Boydas 1 , Bernd Winter 2 , David Batchelor 3 , Michael Roemelt 1
International Journal of Quantum Chemistry ( IF 2.3 ) Pub Date : 2020-10-26 , DOI: 10.1002/qua.26515 Esma Birsen Boydas 1 , Bernd Winter 2 , David Batchelor 3 , Michael Roemelt 1
Affiliation
|
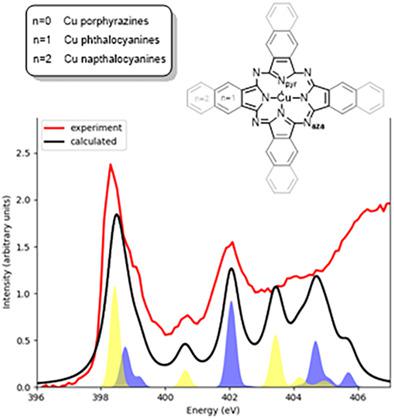
Transition metal porphyrazines are a widely used class of compounds with applications in catalysis, organic solar cells, photodynamic therapy, and nonlinear optics. The most prominent members of that family of compounds are metallophtalocyanines, which have been the subject of numerous spectroscopic and theoretical studies. In this work, the electronic structure and X‐ray absorption characteristics of three Cu‐porphyrazine derivatives are investigated by means of modern electronic structure theory. More precisely, the experimentally observed N K‐edge and Cu L‐edge features are presented and reproduced by time‐dependent density functional theory, restricted open‐shell configuration interaction, and a restricted active space approach. Where possible, the calculations are used to interpret the observed spectroscopic features in terms of electronic transitions and, furthermore, to connect spectral differences to chemical variations. Part of the discussion of the computational results concerns the impact of various parameters and approximations that are used for the calculations, for example, the choice of active space.
中文翻译:

从电子结构理论洞察铜卟啉的X射线吸收光谱
过渡金属卟啉是一类广泛使用的化合物,可用于催化,有机太阳能电池,光动力疗法和非线性光学。该化合物家族中最杰出的成员是金属酞菁,它已成为许多光谱学和理论研究的主题。在这项工作中,通过现代电子结构理论研究了三种铜卟啉衍生物的电子结构和X射线吸收特性。更准确地说,通过时间相关的密度泛函理论,受限的开壳结构相互作用和受限的活动空间方法,展示和再现了实验观察到的NK-edge和Cu L-edge特征。在可能的情况,该计算用于根据电子跃迁解释观察到的光谱特征,此外,还将光谱差异与化学变化联系起来。关于计算结果的部分讨论涉及用于计算的各种参数和近似值的影响,例如,活动空间的选择。
更新日期:2020-12-23
中文翻译:
从电子结构理论洞察铜卟啉的X射线吸收光谱
过渡金属卟啉是一类广泛使用的化合物,可用于催化,有机太阳能电池,光动力疗法和非线性光学。该化合物家族中最杰出的成员是金属酞菁,它已成为许多光谱学和理论研究的主题。在这项工作中,通过现代电子结构理论研究了三种铜卟啉衍生物的电子结构和X射线吸收特性。更准确地说,通过时间相关的密度泛函理论,受限的开壳结构相互作用和受限的活动空间方法,展示和再现了实验观察到的NK-edge和Cu L-edge特征。在可能的情况,该计算用于根据电子跃迁解释观察到的光谱特征,此外,还将光谱差异与化学变化联系起来。关于计算结果的部分讨论涉及用于计算的各种参数和近似值的影响,例如,活动空间的选择。










































 京公网安备 11010802027423号
京公网安备 11010802027423号