当前位置:
X-MOL 学术
›
Comp. Mater. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Surface segregation in multicomponent high entropy alloys: Atomistic simulations versus a multilayer analytical model
Computational Materials Science ( IF 3.1 ) Pub Date : 2021-02-01 , DOI: 10.1016/j.commatsci.2020.110101 Dominique Chatain , Paul Wynblatt
Computational Materials Science ( IF 3.1 ) Pub Date : 2021-02-01 , DOI: 10.1016/j.commatsci.2020.110101 Dominique Chatain , Paul Wynblatt
|
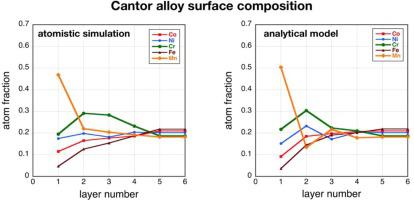
Abstract This paper compares two approaches for investigating the near-surface composition profile that results from surface segregation in the so-called Cantor alloy, an equi-molar alloy of CoCrFeMnNi. One approach consists of atomistic computer simulations by a combination of Monte Carlo, molecular dynamics and molecular statics techniques, and the other is a nearest neighbor analytical calculation performed in the regular solution approximation with a multilayer model, developed here for the first time for a N-component system and tested for the 5-component Cantor alloy. This type of comparison is useful because a typical computer simulation requires the use of ~100 parallel processors for 2 to 3 h, whereas a similar calculation by means of the analytical model can be performed in a few seconds on a laptop machine. The results obtained show qualitatively good agreement between the two approaches. Thus, while the results of the computer simulations are presumably more reliable, and provide an atomic scale picture, if massive computations are required, for example, in order to optimize the composition of a multicomponent alloy, then an initial screening of the composition space by the analytical model could provide a highly useful means of narrowing the regions of interest, in the same way that the CALPHAD method allows rapid investigation of phase diagrams in complex multinary systems.
中文翻译:

多组分高熵合金的表面偏析:原子模拟与多层分析模型
摘要 本文比较了两种研究近表面成分分布的方法,这些方法是由所谓的康托合金(一种 CoCrFeMnNi 的等摩尔合金)中的表面偏析引起的。一种方法包括结合蒙特卡罗、分子动力学和分子静力学技术的原子计算机模拟,另一种方法是使用多层模型在常规解近似中执行的最近邻分析计算,这里首次开发用于 N -组分系统并测试了 5 组分康托合金。这种类型的比较很有用,因为典型的计算机模拟需要使用约 100 个并行处理器持续 2 到 3 小时,而借助分析模型的类似计算可以在几秒钟内在膝上型计算机上执行。获得的结果表明两种方法之间具有良好的定性一致性。因此,虽然计算机模拟的结果可能更可靠,并提供原子尺度的图片,但如果需要大量计算,例如,为了优化多组分合金的成分,则通过以下方式对成分空间进行初步筛选分析模型可以提供一种非常有用的缩小感兴趣区域的方法,就像 CALPHAD 方法允许快速研究复杂多元系统中的相图一样。
更新日期:2021-02-01
中文翻译:
多组分高熵合金的表面偏析:原子模拟与多层分析模型
摘要 本文比较了两种研究近表面成分分布的方法,这些方法是由所谓的康托合金(一种 CoCrFeMnNi 的等摩尔合金)中的表面偏析引起的。一种方法包括结合蒙特卡罗、分子动力学和分子静力学技术的原子计算机模拟,另一种方法是使用多层模型在常规解近似中执行的最近邻分析计算,这里首次开发用于 N -组分系统并测试了 5 组分康托合金。这种类型的比较很有用,因为典型的计算机模拟需要使用约 100 个并行处理器持续 2 到 3 小时,而借助分析模型的类似计算可以在几秒钟内在膝上型计算机上执行。获得的结果表明两种方法之间具有良好的定性一致性。因此,虽然计算机模拟的结果可能更可靠,并提供原子尺度的图片,但如果需要大量计算,例如,为了优化多组分合金的成分,则通过以下方式对成分空间进行初步筛选分析模型可以提供一种非常有用的缩小感兴趣区域的方法,就像 CALPHAD 方法允许快速研究复杂多元系统中的相图一样。










































 京公网安备 11010802027423号
京公网安备 11010802027423号