当前位置:
X-MOL 学术
›
ACS Comb. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Profiling SARS-CoV-2 Main Protease (MPRO) Binding to Repurposed Drugs Using Molecular Dynamics Simulations in Classical and Neural Network-Trained Force Fields
ACS Combinatorial Science Pub Date : 2020-10-29 , DOI: 10.1021/acscombsci.0c00140 Aayush Gupta , Huan-Xiang Zhou
ACS Combinatorial Science Pub Date : 2020-10-29 , DOI: 10.1021/acscombsci.0c00140 Aayush Gupta , Huan-Xiang Zhou
|
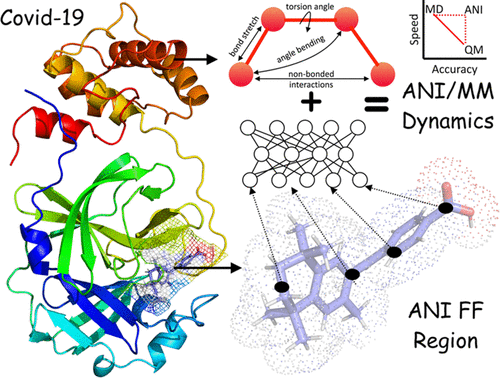
The current COVID-19 pandemic caused by a novel coronavirus SARS-CoV-2 urgently calls for a working therapeutic. Here, we report a computation-based workflow for efficiently selecting a subset of FDA-approved drugs that can potentially bind to the SARS-CoV-2 main protease MPRO. The workflow started with docking (using Autodock Vina) each of 1615 FDA-approved drugs to the MPRO active site. This step selected 62 candidates with docking energies lower than −8.5 kcal/mol. Then, the 62 docked protein–drug complexes were subjected to 100 ns of molecular dynamics (MD) simulations in a molecular mechanics (MM) force field (CHARMM36). This step reduced the candidate pool to 26, based on the root-mean-square-deviations (RMSDs) of the drug molecules in the trajectories. Finally, we modeled the 26 drug molecules by a pseudoquantum mechanical (ANI) force field and ran 5 ns hybrid ANI/MM MD simulations of the 26 protein–drug complexes. ANI was trained by neural network models on quantum mechanical density functional theory (wB97X/6-31G(d)) data points. An RMSD cutoff winnowed down the pool to 12, and free energy analysis (MM/PBSA) produced the final selection of 9 drugs: dihydroergotamine, midostaurin, ziprasidone, etoposide, apixaban, fluorescein, tadalafil, rolapitant, and palbociclib. Of these, three are found to be active in literature reports of experimental studies. To provide physical insight into their mechanism of action, the interactions of the drug molecules with the protein are presented as 2D-interaction maps. These findings and mappings of drug–protein interactions may be potentially used to guide rational drug discovery against COVID-19.
中文翻译:

使用经典和神经网络训练的力场中的分子动力学模拟分析 SARS-CoV-2 主要蛋白酶 (MPRO) 与再利用药物的结合
当前由新型冠状病毒 SARS-CoV-2 引起的 COVID-19 大流行迫切需要有效的治疗方法。在这里,我们报告了一种基于计算的工作流程,用于有效选择 FDA 批准的可能与 SARS-CoV-2 主要蛋白酶 M PRO结合的药物子集。该工作流程首先将 1615 种 FDA 批准的药物分别对接(使用 Autodock Vina)至 M PRO活性位点。此步骤选择了 62 个对接能量低于 -8.5 kcal/mol 的候选者。然后,对 62 个对接的蛋白质-药物复合物在分子力学 (MM) 力场 (CHARMM36) 中进行 100 ns 的分子动力学 (MD) 模拟。此步骤根据轨迹中药物分子的均方根偏差 (RMSD) 将候选池减少到 26 个。最后,我们通过伪量子机械 (ANI) 力场对 26 种药物分子进行建模,并对 26 种蛋白质-药物复合物进行 5 ns 混合 ANI/MM MD 模拟。ANI 通过量子力学密度泛函理论 (wB97X/6-31G(d)) 数据点的神经网络模型进行训练。RMSD 截止值筛选到 12 个,自由能分析 (MM/PBSA) 产生了 9 种药物的最终选择:二氢麦角胺、米斯托林、齐拉西酮、依托泊苷、阿哌沙班、荧光素、他达拉非、罗拉匹坦和哌柏西利。其中,三个被发现在实验研究的文献报告中很活跃。为了提供对其作用机制的物理洞察,药物分子与蛋白质的相互作用以二维相互作用图的形式呈现。这些发现和药物-蛋白质相互作用的图谱可能会用于指导针对 COVID-19 的合理药物发现。
更新日期:2020-12-14
中文翻译:
使用经典和神经网络训练的力场中的分子动力学模拟分析 SARS-CoV-2 主要蛋白酶 (MPRO) 与再利用药物的结合
当前由新型冠状病毒 SARS-CoV-2 引起的 COVID-19 大流行迫切需要有效的治疗方法。在这里,我们报告了一种基于计算的工作流程,用于有效选择 FDA 批准的可能与 SARS-CoV-2 主要蛋白酶 M PRO结合的药物子集。该工作流程首先将 1615 种 FDA 批准的药物分别对接(使用 Autodock Vina)至 M PRO活性位点。此步骤选择了 62 个对接能量低于 -8.5 kcal/mol 的候选者。然后,对 62 个对接的蛋白质-药物复合物在分子力学 (MM) 力场 (CHARMM36) 中进行 100 ns 的分子动力学 (MD) 模拟。此步骤根据轨迹中药物分子的均方根偏差 (RMSD) 将候选池减少到 26 个。最后,我们通过伪量子机械 (ANI) 力场对 26 种药物分子进行建模,并对 26 种蛋白质-药物复合物进行 5 ns 混合 ANI/MM MD 模拟。ANI 通过量子力学密度泛函理论 (wB97X/6-31G(d)) 数据点的神经网络模型进行训练。RMSD 截止值筛选到 12 个,自由能分析 (MM/PBSA) 产生了 9 种药物的最终选择:二氢麦角胺、米斯托林、齐拉西酮、依托泊苷、阿哌沙班、荧光素、他达拉非、罗拉匹坦和哌柏西利。其中,三个被发现在实验研究的文献报告中很活跃。为了提供对其作用机制的物理洞察,药物分子与蛋白质的相互作用以二维相互作用图的形式呈现。这些发现和药物-蛋白质相互作用的图谱可能会用于指导针对 COVID-19 的合理药物发现。










































 京公网安备 11010802027423号
京公网安备 11010802027423号