当前位置:
X-MOL 学术
›
Appl. Surf. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Adsorption of Mg2+ and K+ on the kaolinite (001) surface in aqueous system: A combined DFT and AIMD study with an experimental verification
Applied Surface Science ( IF 6.3 ) Pub Date : 2021-02-01 , DOI: 10.1016/j.apsusc.2020.148158 Zhijun Zhang , Qi Zhou , Ziting Yuan , Liang Zhao , Jianda Dong
Applied Surface Science ( IF 6.3 ) Pub Date : 2021-02-01 , DOI: 10.1016/j.apsusc.2020.148158 Zhijun Zhang , Qi Zhou , Ziting Yuan , Liang Zhao , Jianda Dong
|
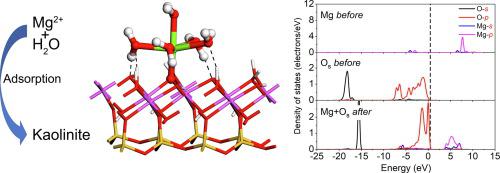
Abstract Microscopic adsorption mechanism of Mg2+ and K+ on kaolinite (0 0 1) surface in aqueous system was investigated by density functional theory (DFT) calculations and ab-initio molecular dynamics (AIMD) simulations. A verification experiment has also been done to compare the adsorption capacity of kaolinite to the two ions. The structure for both hydrated complexes and mono-/bi-dentate adsorption complexes of Mg2+ and K+ in aqueous environment was examined, with radial distribution function and binding energies calculated. [Mg(H2O)6]2+ and [K(H2O)6]+ are the dominant complexes of hydrated Mg2+ and K+, respectively. Bidentate complexes of Mg2+ and monodentate complexes of K+ are their respective dominant complexes, and the binding energies of all the adsorption complexes of Mg2+ are higher than those of K+. Partial density of states projections combining with Mulliken bond charge and populations show the strong ionic characters and weak bonding and anti-bonding states filling of Mg − Os and K − Os in the adsorption complexes. The adsorption experiment verified that Mg2+ is easier to adsorb on kaolinite surface than K+.
中文翻译:

Mg2+ 和 K+ 在水性体系中高岭石 (001) 表面的吸附:结合 DFT 和 AIMD 研究以及实验验证
摘要 通过密度泛函理论(DFT)计算和从头算分子动力学(AIMD)模拟研究了Mg2+和K+在水体系中高岭石(0 0 1)表面的微观吸附机理。还进行了验证实验,比较了高岭石对两种离子的吸附能力。研究了水环境中 Mg2+ 和 K+ 的水合配合物和单齿/双齿吸附配合物的结构,并计算了径向分布函数和结合能。[Mg(H2O)6]2+ 和 [K(H2O)6]+ 分别是水合 Mg2+ 和 K+ 的主要配合物。Mg2+的双齿配合物和K+的单齿配合物是它们各自的优势配合物,所有Mg2+的吸附配合物的结合能都高于K+的结合能。结合Mulliken键电荷和布居的部分态密度投影显示吸附复合物中Mg - Os和K - Os的强离子特性和弱键合和反键合状态填充。吸附实验证实Mg2+比K+更容易吸附在高岭石表面。
更新日期:2021-02-01
中文翻译:
Mg2+ 和 K+ 在水性体系中高岭石 (001) 表面的吸附:结合 DFT 和 AIMD 研究以及实验验证
摘要 通过密度泛函理论(DFT)计算和从头算分子动力学(AIMD)模拟研究了Mg2+和K+在水体系中高岭石(0 0 1)表面的微观吸附机理。还进行了验证实验,比较了高岭石对两种离子的吸附能力。研究了水环境中 Mg2+ 和 K+ 的水合配合物和单齿/双齿吸附配合物的结构,并计算了径向分布函数和结合能。[Mg(H2O)6]2+ 和 [K(H2O)6]+ 分别是水合 Mg2+ 和 K+ 的主要配合物。Mg2+的双齿配合物和K+的单齿配合物是它们各自的优势配合物,所有Mg2+的吸附配合物的结合能都高于K+的结合能。结合Mulliken键电荷和布居的部分态密度投影显示吸附复合物中Mg - Os和K - Os的强离子特性和弱键合和反键合状态填充。吸附实验证实Mg2+比K+更容易吸附在高岭石表面。










































 京公网安备 11010802027423号
京公网安备 11010802027423号