Molecular Cell ( IF 14.5 ) Pub Date : 2020-10-16 , DOI: 10.1016/j.molcel.2020.09.025 Vasilios Zachariadis , Huaitao Cheng , Nathanael Andrews , Martin Enge
|
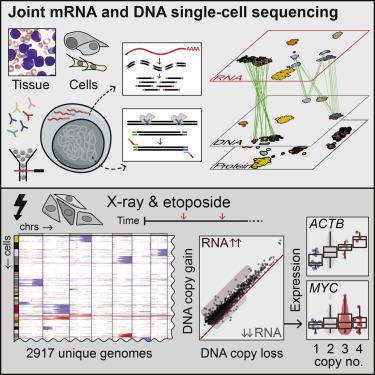
To address how genetic variation alters gene expression in complex cell mixtures, we developed direct nuclear tagmentation and RNA sequencing (DNTR-seq), which enables whole-genome and mRNA sequencing jointly in single cells. DNTR-seq readily identified minor subclones within leukemia patients. In a large-scale DNA damage screen, DNTR-seq was used to detect regions under purifying selection and identified genes where mRNA abundance was resistant to copy-number alteration, suggesting strong genetic compensation. mRNA sequencing (mRNA-seq) quality equals RNA-only methods, and the low positional bias of genomic libraries allowed detection of sub-megabase aberrations at ultra-low coverage. Each cell library is individually addressable and can be re-sequenced at increased depth, allowing multi-tiered study designs. Additionally, the direct tagmentation protocol enables coverage-independent estimation of ploidy, which can be used to identify cell singlets. Thus, DNTR-seq directly links each cell’s state to its corresponding genome at scale, enabling routine analysis of heterogeneous tumors and other complex tissues.
中文翻译:
单细胞联合全基因组测序和基因表达谱分析的高度可扩展方法
为了解决遗传变异如何改变复杂细胞混合物中基因表达的问题,我们开发了直接核标签和RNA测序(DNTR-seq),可在单个细胞中共同进行全基因组和mRNA测序。DNTR-seq可以轻松识别出白血病患者中的次要亚克隆。在大规模的DNA损伤筛选中,DNTR-seq用于检测纯化选择下的区域,并鉴定了mRNA丰度可抵抗拷贝数变化的基因,表明具有强大的遗传补偿能力。mRNA测序(mRNA-seq)的质量等同于仅使用RNA的方法,并且基因组文库的低位置偏差允许以超低覆盖率检测亚兆碱基的畸变。每个细胞库都可以单独寻址,并且可以在更深的深度进行重新排序,从而可以进行多层研究设计。另外,直接标记协议可实现非覆盖性的倍性估算,可用于鉴定细胞单重态。因此,DNTR-seq大规模地将每个细胞的状态与其相应的基因组联系起来,从而能够对异质性肿瘤和其他复杂组织进行常规分析。










































 京公网安备 11010802027423号
京公网安备 11010802027423号