Monatshefte für Chemie - Chemical Monthly ( IF 1.7 ) Pub Date : 2020-10-16 , DOI: 10.1007/s00706-020-02680-9 Parimala Devi Duraisamy , Praveena Gopalan , Abiram Angamuthu
|
Abstract
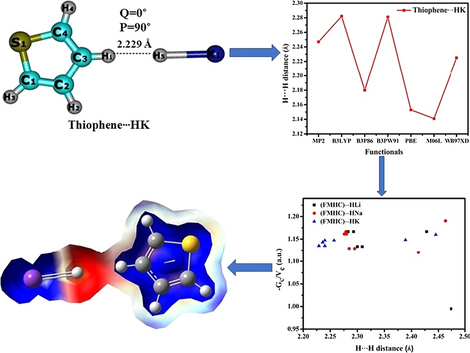
In this work, dihydrogen bond between five-membered heterocyclic compounds (furan, thiophene, pyrrole, arsole, borole, phosphole, and silole) and alkali metal hydride HM (M = Li, Na, and K) were carried out by density functional theory and ab-initio calculations. A two pure (PBE, M06L) and four hybrids (B3LYP, B3P86, B3PW91, wB97XD) DFT functional and MP2 method with 6–311 + + G** basis set were used for five membered heterocyclic dihydrogen bond interactions. Molecular geometries, puckering parameters, energies, and natural bond orbital analysis of all the complexes were calculated by various DFT functionals and MP2 method. For all the complexes, smallest H∙∙∙H bond distance is observed for thiophene∙∙∙HK which has significant interaction energy (∆EC), while largest H∙∙∙H distance with smallest ∆EC is observed for pyrrole∙∙∙HLi complex. Among all the functionals, M06L predicted the smallest dihydrogen bond distances for all the complexes, while largest dihydrogen bond distance was observed by B3LYP functional. The infrared vibrational frequency analysis for all the complexes has revealed propensities in red and blue shift for the C–H and M–H bonds, respectively. Natural bond orbital and quantum theory of atom in molecules analyses were performed to examine the nature of interaction along with the molecular electrostatic potential which further confirm the existence of dihydrogen bonding.
Graphic abstract
中文翻译:
五元杂环配合物中的分子间CH∙∙∙HM二氢键:DFT和从头算研究
摘要
在这项工作中,通过密度泛函理论进行了五元杂环化合物(呋喃,噻吩,吡咯,芳醚,硼,磷和甲硅烷基)与碱金属氢化物HM(M = Li,Na和K)之间的二氢键和ab-initio计算。具有6–311 + + G **基集的两个纯(PBE,M06L)和四个杂种(B3LYP,B3P86,B3PW91,wB97XD)DFT功能和MP2方法用于五元杂环二氢键相互作用。通过各种DFT功能和MP2方法计算了所有配合物的分子几何结构,起皱参数,能量和自然键轨道分析。对于所有配合物,噻吩∙∙∙HK的相互作用能量(∆ E C)都观察到最小的H∙∙∙H键距,而Δ∆最小的H∙∙∙H距离最大吡咯∙∙∙HLi络合物观察到E C。在所有官能团中,M06L预测所有配合物的最小二氢键距离,而通过B3LYP官能团观察到最大的二氢键距离。所有配合物的红外振动频率分析均显示出C–H和M–H键分别发生红移和蓝移。进行了分子中原子的自然键轨道和量子理论分析,以检验相互作用的性质以及分子静电势,这进一步证实了二氢键的存在。









































 京公网安备 11010802027423号
京公网安备 11010802027423号