Journal of Organometallic Chemistry ( IF 2.1 ) Pub Date : 2020-10-14 , DOI: 10.1016/j.jorganchem.2020.121580 Ming-Xing Zhang , Guorui Zuo , Xiaofei Yang , Sheng Hua Liu
|
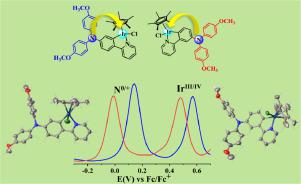
We present herein the synthesis, structure, redox and electronic absorption studies of a series of cyclometalated iridium-triarylamine complexes 1−7. For complexes 1-4 where the amine nitrogen is in the meta position to the Ir–CC^N bond, the electronic nature of the triarylamine units is varied by using different substituents (H, Me, OMe and Cl). In order to study the relationship between the connection mode and the intramolecular electron coupling properties, the amine nitrogen is placed in the para position with respect to the Ir–CC^N bond in complexes 5 and 6. And it should be noted that all of these complexes exhibit two well-defined redox processes (N/N•+ and IrIII/IV) within the available potential window at appreciably low potentials. The neutral, one-electron-oxidized states of complexes 3 and 6 were investigated by cyclic voltammetry (CV) and square-wave voltammetry (SWV), UV-vis-NIR spectroelectrochemistry and theoretical calculations. Experimental results show that the above mentioned four different substituents does not have a significant effect on the potential splitting ΔE between the two anodic redox waves of complexes 1-4. But, the ΔE of complex 6 is larger than that of complex 3, indicating that the electron coupling of arylamine and iridium units in the para position (6) is stronger than that in the meta position (3), which is also verified by in-situ absorption spectroelectrochemistry. It is worth noting that the directions of intramolecular charge-transfer in complexes 1 and 5 is reversed due to the different arylamine groups attachment sites (either meta or para to the Ir–CC^N bond).
中文翻译:
芳基氨基修饰的环金属化铱(III)配合物的合成和氧化还原性能
我们在此介绍一系列环金属化铱-三芳基胺络合物1-7的合成,结构,氧化还原和电子吸收研究。对于其中胺氮位于Ir–C C ^ N键间位的配合物1-4,三芳基胺单元的电子性质通过使用不同的取代基(H,Me,OMe和Cl)而变化。为了研究连接方式与分子内电子偶联性质之间的关系,将胺氮相对于配合物5和6中的Ir–C C ^ N键置于对位。应该指出的是,所有这些络合物都表现出两个定义明确的氧化还原过程(N / N •+和Ir III / IV)在相当低的电位范围内。通过循环伏安法(CV)和方波伏安法(SWV),UV-vis-NIR光谱电化学和理论计算研究了配合物3和6的中性,一电子氧化态。实验结果表明,上述四个不同的取代基对配合物1-4的两个阳极氧化还原波之间的电位分裂ΔE没有显着影响。但是,配合物6的ΔE大于配合物3的ΔE ,表明芳基胺和铱单元在对位(6)比间位(3)更强,这也通过原位吸收光谱电化学证实。值得注意的是,由于不同的芳胺基团附着位点(Ir-CC C ^ N键的间位或对位),复合物1和5中分子内电荷转移的方向相反。










































 京公网安备 11010802027423号
京公网安备 11010802027423号