当前位置:
X-MOL 学术
›
Clin. Genet.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Missing heritability in Bloom syndrome: First report of a deep intronic variant leading to pseudo‐exon activation in the BLM gene
Clinical Genetics ( IF 2.9 ) Pub Date : 2020-10-09 , DOI: 10.1111/cge.13859 Lynn Backers 1, 2 , Bram Parton 1, 2 , Marieke De Bruyne 1 , Simon J Tavernier 3, 4 , Kris Van Den Bogaert 5 , Bart N Lambrecht 6, 7 , Filomeen Haerynck 7 , Kathleen B M Claes 1, 2
Clinical Genetics ( IF 2.9 ) Pub Date : 2020-10-09 , DOI: 10.1111/cge.13859 Lynn Backers 1, 2 , Bram Parton 1, 2 , Marieke De Bruyne 1 , Simon J Tavernier 3, 4 , Kris Van Den Bogaert 5 , Bart N Lambrecht 6, 7 , Filomeen Haerynck 7 , Kathleen B M Claes 1, 2
Affiliation
|
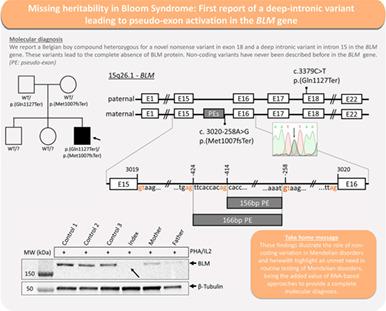
Pathogenic biallelic variants in the BLM/RECQL3 gene cause a rare autosomal recessive disorder called Bloom syndrome (BS). This syndrome is characterized by severe growth delay, immunodeficiency, dermatological manifestations and a predisposition to a wide variety of cancers, often multiple and very early in life. Literature shows that the main mode of BLM inactivation is protein translation termination. We expanded the molecular spectrum of BS by reporting the first deep intronic variant causing intron exonisation. We describe a patient with a clinical phenotype of BS and a strong increase in sister chromatid exchanges (SCE), who was found to be compound heterozygous for a novel nonsense variant c.3379C>T, p.(Gln1127Ter) in exon 18 and a deep intronic variant c.3020‐258A>G in intron 15 of the BLM gene. The deep intronic variant creates a high‐quality de novo donor splice site, which leads to retention of two intron segments. Both pseudo‐exons introduce a premature stop codon into the reading frame and abolish BLM protein expression, confirmed by Western Blot analysis. These findings illustrate the role of non‐coding variation in Mendelian disorders and herewith highlight an unmet need in routine testing of Mendelian disorders, being the added value of RNA‐based approaches to provide a complete molecular diagnosis.
中文翻译:

Bloom 综合征遗传力缺失:导致 BLM 基因假外显子激活的深度内含子变异的首次报告
BLM/RECQL3基因中的致病性双等位基因变异导致一种罕见的常染色体隐性遗传病,称为 Bloom 综合征 (BS)。这种综合征的特点是严重的生长迟缓、免疫缺陷、皮肤病表现和多种癌症的易感性,通常是多种癌症,而且很早。文献表明,BLM 失活的主要方式是蛋白质翻译终止。我们通过报告第一个导致内含子外显子化的深度内含子变异扩展了 BS 的分子谱。我们描述了一名具有 BS 临床表型和姐妹染色单体交换 (SCE) 显着增加的患者,该患者被发现是外显子 18 和 a BLM内含子 15 中的深内含子变异 c.3020-258A>G基因。深内含子变体创建了一个高质量的从头供体剪接位点,从而保留了两个内含子片段。通过蛋白质印迹分析证实,两个假外显子都将提前终止密码子引入阅读框并消除 BLM 蛋白表达。这些发现说明了非编码变异在孟德尔疾病中的作用,因此强调了孟德尔疾病常规检测中未满足的需求,这是基于 RNA 的方法提供完整分子诊断的附加价值。
更新日期:2020-10-09
中文翻译:
Bloom 综合征遗传力缺失:导致 BLM 基因假外显子激活的深度内含子变异的首次报告
BLM/RECQL3基因中的致病性双等位基因变异导致一种罕见的常染色体隐性遗传病,称为 Bloom 综合征 (BS)。这种综合征的特点是严重的生长迟缓、免疫缺陷、皮肤病表现和多种癌症的易感性,通常是多种癌症,而且很早。文献表明,BLM 失活的主要方式是蛋白质翻译终止。我们通过报告第一个导致内含子外显子化的深度内含子变异扩展了 BS 的分子谱。我们描述了一名具有 BS 临床表型和姐妹染色单体交换 (SCE) 显着增加的患者,该患者被发现是外显子 18 和 a BLM内含子 15 中的深内含子变异 c.3020-258A>G基因。深内含子变体创建了一个高质量的从头供体剪接位点,从而保留了两个内含子片段。通过蛋白质印迹分析证实,两个假外显子都将提前终止密码子引入阅读框并消除 BLM 蛋白表达。这些发现说明了非编码变异在孟德尔疾病中的作用,因此强调了孟德尔疾病常规检测中未满足的需求,这是基于 RNA 的方法提供完整分子诊断的附加价值。









































 京公网安备 11010802027423号
京公网安备 11010802027423号