Physica E: Low-dimensional Systems and Nanostructures ( IF 2.9 ) Pub Date : 2020-10-09 , DOI: 10.1016/j.physe.2020.114472 Naidel A.M.S. Caturello , Rafael Besse , Julian F.R.V. Silveira , Matheus P. Lima , Juarez L.F. Da Silva
|
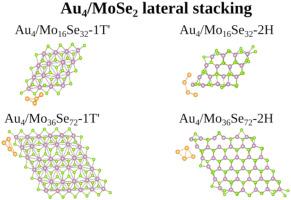
An atomistic understanding of the interaction between Au clusters and dichalcogenides nanoflakes of different polytypes is fundamental to improve our knowledge to the tuning of the physical-chemical properties of hybrid dimensional composite materials. Here, we report a density functional theory investigation into the changes of structural, energetic, and electronic properties induced by the adsorption of Au4 clusters (planar and tetrahedron) onto Mo16Se32 and Mo36Se72 nanoflakes of both 1T’ and 2H polytypes. We found that the Au–Se interaction plays a critical role in the nature of the Au4/MoSe2 interactions, in which there is a strong energetic preference for the S atoms located at the edges for all nanoflake sizes and polytypes. In summary, the Au⋯MoSe2 binding mechanisms is composed of either (i) splitting of Se sp2-orbitals interacting with Au sd3-orbitals, or (ii) re-hybridized Au sd3 splitting of Mo -orbitals, with low charge transfer in both due to Au4 cluster electron filling. Furthermore, the cluster/nanoflake adsorption energetics and structural distortions due to the Au⋯MoSe2 interactions are mainly determined by the MoSe2 edge configurations.
中文翻译:
第一原理洞察边缘在MoSe 2纳米薄片上Au 4团簇的结合机理中的作用
对Au团簇和不同多型二卤化碳纳米薄片之间相互作用的原子学理解,对于提高我们对混合尺寸复合材料的物理化学性质进行调节的知识至关重要。在这里,我们报告密度泛函理论研究的结构,能量和电子性质的变化,由Au 4团簇(平面和四面体)吸附到1T'和Mo的Mo 16 Se 32和Mo 3 6Se 7 2纳米薄片上引起的2H多型。我们发现Au-Se相互作用在Au 4 / MoSe 2的性质中起着至关重要的作用相互作用,其中对于所有纳米片大小和多型体,位于边缘的S原子都有强烈的能量偏好。总之,在Au⋯摩西2结合机构是由任一(的我的Se)分裂SP 2个-orbitals用Au相互作用SD 3个-orbitals,或(II)重新杂交的Au SD 3分割的Mo-轨道,由于Au 4团簇电子填充,电荷转移都很低。此外,由于Au⋯MoSe 2相互作用引起的团簇/纳米片状吸附能和结构变形主要由MoSe 2边缘构型决定。










































 京公网安备 11010802027423号
京公网安备 11010802027423号