当前位置:
X-MOL 学术
›
Bull. Korean Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Density Functional Theory Calculations of the Adsorption of Cytosine on Si(100)
Bulletin of the Korean Chemical Society ( IF 2.3 ) Pub Date : 2020-10-07 , DOI: 10.1002/bkcs.12108 Hyun‐Kyung Kim 1 , Do Hwan Kim 1
Bulletin of the Korean Chemical Society ( IF 2.3 ) Pub Date : 2020-10-07 , DOI: 10.1002/bkcs.12108 Hyun‐Kyung Kim 1 , Do Hwan Kim 1
Affiliation
|
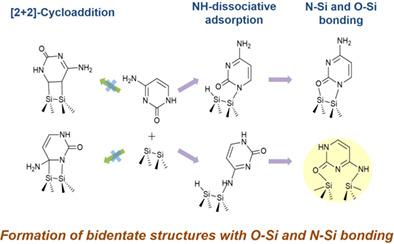
We have performed density functional theory (DFT) calculations to investigate the adsorption structures of cytosine on Si(100). Multiple adsorption configurations that could result from Lewis acid–base reactions or cycloadditions were optimized, and the structural and energetic parameters of the obtained adsorption structures were compared. Row‐bridged bidentate configurations with SiOCNCNSi linkages to two silicon atoms in adjacent dimer rows were found to be more stable than other configurations. The SiN bond was formed by NH dissociative adsorption, while the SiO bond was dative. In the monodentate structures, the CH or NH dissociative adsorption structures were more energetically favorable than the datively bonded ones. Cycloaddition through the π‐electrons of the CN or CC bonds of the cytosine ring resulted in relatively unstable adsorption products.
中文翻译:

胞嘧啶在Si(100)上的吸附的密度泛函理论计算
我们已经进行了密度泛函理论(DFT)计算,以研究胞嘧啶在Si(100)上的吸附结构。对路易斯酸碱反应或环加成反应可能产生的多种吸附构型进行了优化,并对得到的吸附结构的结构和能级参数进行了比较。有Si行桥接的二齿配置 ö Ç Ñ Ç Ñ Si键,以在相邻的二聚体排中的两个硅原子被发现是比其它构造更稳定。在Si N键是由N个形成 ħ解离吸附,而Si O键是固定的。在单齿结构中,C H或ñ ħ解离吸附结构均高于datively粘结那些更积极有利的。通过胞嘧啶环的CN或CC键的π电子进行环加成反应会产生相对不稳定的吸附产物。
更新日期:2020-11-13
中文翻译:
胞嘧啶在Si(100)上的吸附的密度泛函理论计算
我们已经进行了密度泛函理论(DFT)计算,以研究胞嘧啶在Si(100)上的吸附结构。对路易斯酸碱反应或环加成反应可能产生的多种吸附构型进行了优化,并对得到的吸附结构的结构和能级参数进行了比较。有Si行桥接的二齿配置 ö Ç Ñ Ç Ñ Si键,以在相邻的二聚体排中的两个硅原子被发现是比其它构造更稳定。在Si N键是由N个形成 ħ解离吸附,而Si O键是固定的。在单齿结构中,C H或ñ ħ解离吸附结构均高于datively粘结那些更积极有利的。通过胞嘧啶环的CN或CC键的π电子进行环加成反应会产生相对不稳定的吸附产物。








































 京公网安备 11010802027423号
京公网安备 11010802027423号