当前位置:
X-MOL 学术
›
Birth Defects Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Separation in genetic pathogenesis of mutations in FBN1‐TB5 region between autosomal dominant acromelic dysplasia and Marfan syndrome
Birth Defects Research ( IF 1.6 ) Pub Date : 2020-10-08 , DOI: 10.1002/bdr2.1814 Chengjun Sun 1 , Dandan Xu 1 , Zhou Pei 1 , Lin Yang 1, 2 , Zhongwei Qiao 3 , Wei Lu 1 , Feihong Luo 1 , Zhengqing Qiu 4
Birth Defects Research ( IF 1.6 ) Pub Date : 2020-10-08 , DOI: 10.1002/bdr2.1814 Chengjun Sun 1 , Dandan Xu 1 , Zhou Pei 1 , Lin Yang 1, 2 , Zhongwei Qiao 3 , Wei Lu 1 , Feihong Luo 1 , Zhengqing Qiu 4
Affiliation
|
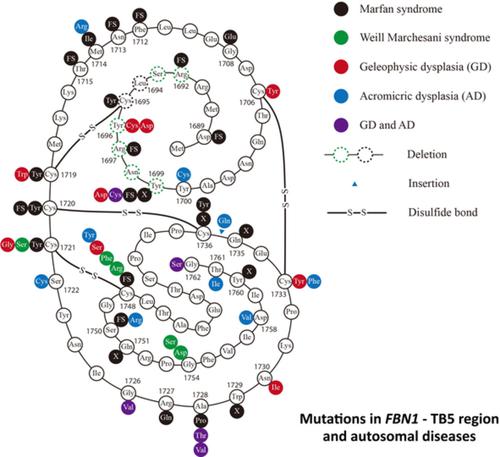
Mutations in the transforming growth factor β‐binding protein‐like domain 5 (TB5) region of FBN1 can lead to autosomal acromelic dysplasia and Marfan syndrome, which are two diseases with apparently opposite phenotypes. We identified six patients with acromelic dysplasia carrying either the previously reported mutations c.5284G > A (p.Gly1762Ser) and c.5096A > G (p.Tyr1699Cys) or the novel mutation c.5260G > A (p.Gly1754Ser). A systematic review of patients with mutations in the FBN1‐TB5 region showed that acromelic dysplasia is caused only by in‐frame amino acid substitutions. In contrast, truncating mutations in the FBN1‐TB5 have been reported only in Marfan syndrome. Acromelic dysplasia subtypes that share symptoms with Marfan syndrome are associated with FBN1‐TB5 disulfide disruptions, which are also commonly found in Marfan syndrome. These results suggest that the type and location of mutations in the FBN1‐TB5 region determine the clinical spectrum of fibrillinopathy.
中文翻译:

常染色体显性肢端发育不良与马凡综合征FBN1-TB5区突变的遗传发病机制分离
FBN1的转化生长因子β结合蛋白样结构域5(TB5)区域的突变可导致常染色体肢端畸形和马凡综合征,这是两种表型明显相反的疾病。我们确定了 6 名患有肢端发育不良的患者,其携带先前报告的突变 c.5284G > A (p.Gly1762Ser) 和 c.5096A > G (p.Tyr1699Cys) 或新突变 c.5260G > A (p.Gly1754Ser)。对FBN1- TB5 区域突变患者的系统评价表明,肢端发育不良仅由框内氨基酸取代引起。相比之下,仅在马凡综合征中报道了FBN1- TB5中的截断突变。与马凡综合征共有症状的肢端发育不良亚型与FBN1 ‐TB5 二硫键破坏,这在马凡综合征中也很常见。这些结果表明FBN1- TB5 区域突变的类型和位置决定了纤维蛋白病的临床谱。
更新日期:2020-12-01
中文翻译:
常染色体显性肢端发育不良与马凡综合征FBN1-TB5区突变的遗传发病机制分离
FBN1的转化生长因子β结合蛋白样结构域5(TB5)区域的突变可导致常染色体肢端畸形和马凡综合征,这是两种表型明显相反的疾病。我们确定了 6 名患有肢端发育不良的患者,其携带先前报告的突变 c.5284G > A (p.Gly1762Ser) 和 c.5096A > G (p.Tyr1699Cys) 或新突变 c.5260G > A (p.Gly1754Ser)。对FBN1- TB5 区域突变患者的系统评价表明,肢端发育不良仅由框内氨基酸取代引起。相比之下,仅在马凡综合征中报道了FBN1- TB5中的截断突变。与马凡综合征共有症状的肢端发育不良亚型与FBN1 ‐TB5 二硫键破坏,这在马凡综合征中也很常见。这些结果表明FBN1- TB5 区域突变的类型和位置决定了纤维蛋白病的临床谱。










































 京公网安备 11010802027423号
京公网安备 11010802027423号