Journal of Molecular Graphics and Modelling ( IF 2.7 ) Pub Date : 2020-10-08 , DOI: 10.1016/j.jmgm.2020.107772 Marisol Ibarra-Rodríguez 1 , Mario Sánchez 1
|
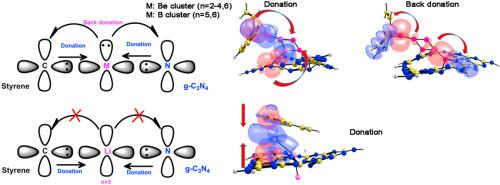
The adsorption of boron, beryllium and lithium clusters on graphitic carbon nitride g-C3N4, and the adsorption of styrene molecule on the B, Be, Li cluster/g-C3N4 sheet have been investigated through the density functional theory (DFT) calculations. Our calculations show distortion of the geometry of the clusters when coordinating with the g-C3N4 sheet. Boron (n = 5 and 6), beryllium (n = 2–4, 6) and Li3 cluster on g-C3N4 present characteristics to adsorb a styrene molecule. The styrene on Be4/g-C3N4 system exhibits better adsorption, due to the beryllium atoms have strong interactions with the π-orbitals of the aromatic ring of the styrene molecule. The study of natural bond orbitals of styrene-cluster/g-C3N4 systems showed the donation process from the styrene molecule and the g-C3N4 sheet towards the boron, beryllium and lithium clusters. Only back donation was observed the boron and beryllium clusters.
中文翻译:
硼,铍和锂团簇(n = 2–6)的理论研究,在石墨氮化碳上的吸附以及[cluster / gC 3 N 4 ]系统中苯乙烯分子配位的受体-供体轨道的研究
通过密度泛函理论(DFT)计算研究了石墨氮化碳gC 3 N 4上硼,铍和锂团簇的吸附以及B,Be,Li簇/ gC 3 N 4板上苯乙烯分子的吸附。。我们的计算结果表明,与gC 3 N 4板材配合时,团簇的几何形状发生了变形。gC 3 N 4上的硼(n = 5和6),铍(n = 2-4、6)和Li 3团簇具有吸附苯乙烯分子的特性。Be 4 / gC 3 N 4上的苯乙烯由于铍原子与苯乙烯分子芳环的π-轨道具有很强的相互作用,因此该体系具有更好的吸附性。对苯乙烯-簇/ gC 3 N 4体系的自然键轨道的研究表明,苯乙烯分子和gC 3 N 4薄层向硼,铍和锂簇的捐赠过程。仅观察到背捐赠的硼和铍簇。










































 京公网安备 11010802027423号
京公网安备 11010802027423号