当前位置:
X-MOL 学术
›
J. Proteome Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Molecular Simulations and Network Modeling Reveal an Allosteric Signaling in the SARS-CoV-2 Spike Proteins
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2020-10-02 , DOI: 10.1021/acs.jproteome.0c00654 Gennady M Verkhivker 1, 2
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2020-10-02 , DOI: 10.1021/acs.jproteome.0c00654 Gennady M Verkhivker 1, 2
Affiliation
|
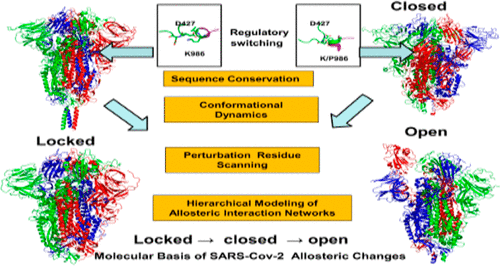
The development of computational strategies for the quantitative characterization of the functional mechanisms of SARS-CoV-2 spike proteins is of paramount importance in efforts to accelerate the discovery of novel therapeutic agents and vaccines combating the COVID-19 pandemic. Structural and biophysical studies have recently characterized the conformational landscapes of the SARS-CoV-2 spike glycoproteins in the prefusion form, revealing a spectrum of stable and more dynamic states. By employing molecular simulations and network modeling approaches, this study systematically examined functional dynamics and identified the regulatory centers of allosteric interactions for distinct functional states of the wild-type and mutant variants of the SARS-CoV-2 prefusion spike trimer. This study presents evidence that the SARS-CoV-2 spike protein can function as an allosteric regulatory engine that fluctuates between dynamically distinct functional states. Perturbation-based modeling of the interaction networks revealed a key role of the cross-talk between the effector hotspots in the receptor binding domain and the fusion peptide proximal region of the SARS-CoV-2 spike protein. The results have shown that the allosteric hotspots of the interaction networks in the SARS-CoV-2 spike protein can control the dynamic switching between functional conformational states that are associated with virus entry to the host receptor. This study offers a useful and novel perspective on the underlying mechanisms of the SARS-CoV-2 spike protein through the lens of allosteric signaling as a regulatory apparatus of virus transmission that could open up opportunities for targeted allosteric drug discovery against SARS-CoV-2 proteins and contribute to the rapid response to the current and potential future pandemic scenarios.
中文翻译:

分子模拟和网络建模揭示了 SARS-CoV-2 刺突蛋白中的变构信号传导
开发用于定量表征 SARS-CoV-2 刺突蛋白功能机制的计算策略对于加速发现抗击 COVID-19 大流行的新型治疗剂和疫苗至关重要。结构和生物物理学研究最近描述了预融合形式的 SARS-CoV-2 刺突糖蛋白的构象景观,揭示了一系列稳定和更动态的状态。通过采用分子模拟和网络建模方法,本研究系统地检查了功能动力学,并确定了 SARS-CoV-2 融合前刺突三聚体野生型和突变体不同功能状态的变构相互作用的调节中心。这项研究提供的证据表明,SARS-CoV-2 刺突蛋白可以作为变构调节引擎,在动态不同的功能状态之间波动。基于扰动的相互作用网络建模揭示了受体结合域中的效应器热点与 SARS-CoV-2 刺突蛋白的融合肽近端区域之间串扰的关键作用。结果表明,SARS-CoV-2刺突蛋白中相互作用网络的变构热点可以控制与病毒进入宿主受体相关的功能构象状态之间的动态切换。这项研究通过变构信号作为病毒传播的调节装置,为 SARS-CoV-2 刺突蛋白的潜在机制提供了有用且新颖的视角,这可能为针对 SARS-CoV-2 的靶向变构药物发现提供机会蛋白质并有助于快速应对当前和未来潜在的大流行情况。
更新日期:2020-11-06
中文翻译:
分子模拟和网络建模揭示了 SARS-CoV-2 刺突蛋白中的变构信号传导
开发用于定量表征 SARS-CoV-2 刺突蛋白功能机制的计算策略对于加速发现抗击 COVID-19 大流行的新型治疗剂和疫苗至关重要。结构和生物物理学研究最近描述了预融合形式的 SARS-CoV-2 刺突糖蛋白的构象景观,揭示了一系列稳定和更动态的状态。通过采用分子模拟和网络建模方法,本研究系统地检查了功能动力学,并确定了 SARS-CoV-2 融合前刺突三聚体野生型和突变体不同功能状态的变构相互作用的调节中心。这项研究提供的证据表明,SARS-CoV-2 刺突蛋白可以作为变构调节引擎,在动态不同的功能状态之间波动。基于扰动的相互作用网络建模揭示了受体结合域中的效应器热点与 SARS-CoV-2 刺突蛋白的融合肽近端区域之间串扰的关键作用。结果表明,SARS-CoV-2刺突蛋白中相互作用网络的变构热点可以控制与病毒进入宿主受体相关的功能构象状态之间的动态切换。这项研究通过变构信号作为病毒传播的调节装置,为 SARS-CoV-2 刺突蛋白的潜在机制提供了有用且新颖的视角,这可能为针对 SARS-CoV-2 的靶向变构药物发现提供机会蛋白质并有助于快速应对当前和未来潜在的大流行情况。










































 京公网安备 11010802027423号
京公网安备 11010802027423号