当前位置:
X-MOL 学术
›
Clin. Genet.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Copy number alterations involving 59 ACMG‐recommended secondary findings genes
Clinical Genetics ( IF 2.9 ) Pub Date : 2020-10-03 , DOI: 10.1111/cge.13852 Svetlana A Yatsenko 1, 2, 3, 4 , Mahmoud Aarabi 2 , Jie Hu 2 , Urvashi Surti 1 , Damara Ortiz 5 , Suneeta Madan-Khetarpal 5 , Devereux N Saller 2 , Daniel Bellissimo 2 , Aleksandar Rajkovic 6, 7
Clinical Genetics ( IF 2.9 ) Pub Date : 2020-10-03 , DOI: 10.1111/cge.13852 Svetlana A Yatsenko 1, 2, 3, 4 , Mahmoud Aarabi 2 , Jie Hu 2 , Urvashi Surti 1 , Damara Ortiz 5 , Suneeta Madan-Khetarpal 5 , Devereux N Saller 2 , Daniel Bellissimo 2 , Aleksandar Rajkovic 6, 7
Affiliation
|
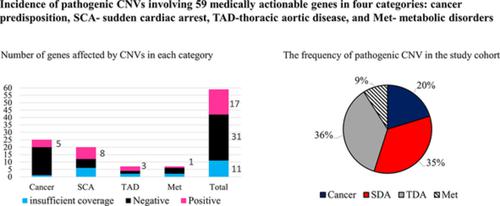
In clinical exome/genome sequencing, the American College of Medical Genetics and Genomics (ACMG) recommends reporting of secondary findings unrelated to a patient's phenotype when pathogenic single‐nucleotide variants (SNVs) are observed in one of 59 genes associated with a life‐threatening, medically actionable condition. Little is known about the incidence and sensitivity of chromosomal microarray analysis (CMA) for detection of pathogenic copy number variants (CNVs) comprising medically‐actionable genes. Clinical CMA has been performed on 8865 individuals referred for molecular cytogenetic testing. We retrospectively reviewed the CMA results to identify patients with CNVs comprising genes included in the 59‐ACMG list of secondary findings. We evaluated the clinical significance of these CNVs in respect to pathogenicity, phenotypic manifestations, and heritability. We identified 23 patients (0.26%) with relevant CNV either deletions comprising the entire gene or intragenic alterations involving one or more secondary findings genes. A number of patients and/or their family members with pathogenic CNVs manifest or expected to develop an anticipated clinical phenotype and would benefit from preventive management similar to the patients with pathogenic SNVs. To improve patients' care standardization should apply to reporting of both sequencing and CNVs obtained via clinical genome‐wide analysis, including chromosomal microarray and exome/genome sequencing.
中文翻译:

涉及 59 个 ACMG 推荐的次要发现基因的拷贝数改变
在临床外显子组/基因组测序中,当在与危及生命相关的 59 个基因之一中观察到致病性单核苷酸变异 (SNV) 时,美国医学遗传学和基因组学学院 (ACMG) 建议报告与患者表型无关的次要发现。 , 医学上可行的条件。关于染色体微阵列分析 (CMA) 检测包含医学可行基因的致病性拷贝数变异 (CNV) 的发生率和敏感性知之甚少。已对 8865 名转诊进行分子细胞遗传学检测的个体进行了临床 CMA。我们回顾性地审查了 CMA 结果,以确定包含 59-ACMG 次要发现列表中包含的基因的 CNV 患者。我们评估了这些 CNV 在致病性方面的临床意义,表型表现和遗传性。我们确定了 23 名患者 (0.26%) 具有相关的 CNV,要么是包含整个基因的缺失,要么是涉及一个或多个次要发现基因的基因内改变。许多具有致病性 CNV 的患者和/或其家庭成员表现出或预期会发展出预期的临床表型,并且将受益于类似于具有致病性 SNV 的患者的预防性管理。为了提高患者的护理标准化,应适用于通过临床全基因组分析(包括染色体微阵列和外显子组/基因组测序)获得的测序和 CNV 的报告。许多具有致病性 CNV 的患者和/或其家庭成员表现出或预期会发展出预期的临床表型,并且将受益于类似于具有致病性 SNV 的患者的预防性管理。为了提高患者的护理标准化,应适用于通过临床全基因组分析(包括染色体微阵列和外显子组/基因组测序)获得的测序和 CNV 的报告。许多具有致病性 CNV 的患者和/或其家庭成员表现出或预期会发展出预期的临床表型,并且将受益于类似于具有致病性 SNV 的患者的预防性管理。为了提高患者的护理标准化,应适用于通过临床全基因组分析(包括染色体微阵列和外显子组/基因组测序)获得的测序和 CNV 的报告。
更新日期:2020-11-18
中文翻译:
涉及 59 个 ACMG 推荐的次要发现基因的拷贝数改变
在临床外显子组/基因组测序中,当在与危及生命相关的 59 个基因之一中观察到致病性单核苷酸变异 (SNV) 时,美国医学遗传学和基因组学学院 (ACMG) 建议报告与患者表型无关的次要发现。 , 医学上可行的条件。关于染色体微阵列分析 (CMA) 检测包含医学可行基因的致病性拷贝数变异 (CNV) 的发生率和敏感性知之甚少。已对 8865 名转诊进行分子细胞遗传学检测的个体进行了临床 CMA。我们回顾性地审查了 CMA 结果,以确定包含 59-ACMG 次要发现列表中包含的基因的 CNV 患者。我们评估了这些 CNV 在致病性方面的临床意义,表型表现和遗传性。我们确定了 23 名患者 (0.26%) 具有相关的 CNV,要么是包含整个基因的缺失,要么是涉及一个或多个次要发现基因的基因内改变。许多具有致病性 CNV 的患者和/或其家庭成员表现出或预期会发展出预期的临床表型,并且将受益于类似于具有致病性 SNV 的患者的预防性管理。为了提高患者的护理标准化,应适用于通过临床全基因组分析(包括染色体微阵列和外显子组/基因组测序)获得的测序和 CNV 的报告。许多具有致病性 CNV 的患者和/或其家庭成员表现出或预期会发展出预期的临床表型,并且将受益于类似于具有致病性 SNV 的患者的预防性管理。为了提高患者的护理标准化,应适用于通过临床全基因组分析(包括染色体微阵列和外显子组/基因组测序)获得的测序和 CNV 的报告。许多具有致病性 CNV 的患者和/或其家庭成员表现出或预期会发展出预期的临床表型,并且将受益于类似于具有致病性 SNV 的患者的预防性管理。为了提高患者的护理标准化,应适用于通过临床全基因组分析(包括染色体微阵列和外显子组/基因组测序)获得的测序和 CNV 的报告。










































 京公网安备 11010802027423号
京公网安备 11010802027423号