Chemical Physics Letters ( IF 2.8 ) Pub Date : 2020-10-01 , DOI: 10.1016/j.cplett.2020.138053 Shuai Zhao , Chunfeng Lan
|
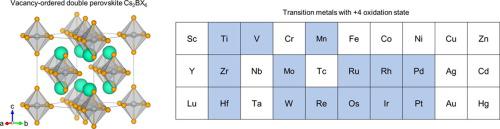
Replacing toxic Pb of halide perovskites is a serious challenge in the current investigation of perovskite solar cells. Vacancy-ordered double halide perovskites have attracted considerable research interest due to their excellent stability and optoelectronic properties. A series of vacancy-ordered double perovskites based on transition metal cations are comprehensively investigated in this work through first-principles calculations. Various electronic configurations of tetravalent transition metals are considered in spin-polarized calculation. Our calculations reveal electronic properties of vacancy-ordered double halide perovskites and provide a theoretical basis for the exploration of novel lead-free perovskites.
中文翻译:
过渡金属基空位有序卤化物钙钛矿电子性质的密度泛函研究
在钙钛矿太阳能电池的当前研究中,替换卤化钙钛矿的有毒铅是一个严峻的挑战。空位有序的双卤化物钙钛矿因其出色的稳定性和光电性能而引起了广泛的研究兴趣。通过第一性原理计算,对基于过渡金属阳离子的一系列空位有序的钙钛矿进行了全面研究。自旋极化计算中考虑了四价过渡金属的各种电子构型。我们的计算揭示了空位有序的双卤化物钙钛矿的电子性质,并为探索新型无铅钙钛矿提供了理论基础。










































 京公网安备 11010802027423号
京公网安备 11010802027423号