当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Equilibrium molecular structure and spectra of 6-methyl-1,5-diazabicyclo[3.1.0]hexane: joint analysis of gas phase electron diffraction, quantum chemistry, and spectroscopic data
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2020-09-30 , DOI: 10.1039/d0cp04005c Leonid S. Khaikin 1, 2, 3, 4 , Georgiy G. Ageev 1, 2, 3, 4 , Anatoliy N. Rykov 1, 2, 3, 4 , Olga E. Grikina 1, 2, 3, 4 , Igor F. Shishkov 1, 2, 3, 4 , Igor V. Kochikov 2, 3, 4, 5 , Vladimir V. Kuznetsov 4, 6, 7, 8 , Nina N. Makhova 4, 6, 7, 8 , Sergei S. Bukalov 3, 4, 7, 9 , Larisa A. Leites 3, 4, 7, 9
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2020-09-30 , DOI: 10.1039/d0cp04005c Leonid S. Khaikin 1, 2, 3, 4 , Georgiy G. Ageev 1, 2, 3, 4 , Anatoliy N. Rykov 1, 2, 3, 4 , Olga E. Grikina 1, 2, 3, 4 , Igor F. Shishkov 1, 2, 3, 4 , Igor V. Kochikov 2, 3, 4, 5 , Vladimir V. Kuznetsov 4, 6, 7, 8 , Nina N. Makhova 4, 6, 7, 8 , Sergei S. Bukalov 3, 4, 7, 9 , Larisa A. Leites 3, 4, 7, 9
Affiliation
|
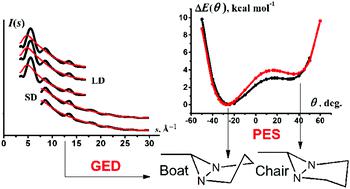
The equilibrium geometry of the boat conformation (Cs point group symmetry) of the 6-methyl-1,5-diazabicyclo[3.1.0]hexane (MDABH) molecule, absolutely dominating under normal conditions, was studied by the gas-phase electron diffraction (GED) method at 20 °C with the involvement of NMR, IR, and Raman spectroscopic data and quantum chemical calculations. The potential function of ring-puckering deformation for the MDABH bicyclic system was calculated at the MP2/aug-cc-pVTZ and B3LYP/cc-pVTZ levels. It was found by MP2 calculation that the total energy of the boat conformation is 3.52 kcal mol−1 lower than that of the chair conformation. For the first time, we recorded the IR and Raman spectra for liquid samples of MDABH and assigned their peculiarities only to boat conformation vibrations using the Pulay technique of scaling quantum chemical force fields. In the case of the chair form, transferability of the refined scale factors was used for reliable prediction of the location of its fundamental frequencies. According to the joint structural analysis of the above data, the most important equilibrium geometric re-parameters for the boat conformation of the MDABH molecule were determined to be (bond lengths in Å; angles in degrees, Cs symmetry): C2N1 = 1.466(2), C2C3 = 1.523(2), N1N5 = 1.512(2), C6N1 = 1.440(2), C6C7 = 1.487(2), ∠C2N1N5 = 106.1(2), ∠N1C2C3) = 110.2(4), ∠C2C3C4 = 99.9(4), ∠N1N5C6 = 58.3(1), ∠N1C6N5 = 63.3(1), ∠N1C6C7 = 114.9(6), ∠C6N1C2 = 111.8(1), ∠N5N1C2C3 = 17.3(1), ∠N1C2C3C4 = −26.8(2), θ = C2C3C4/C2N1N5C4 = −26.2(3), φ = N1C6N5/C2N1N5C4 = 74.0(1). Comparison of these and earlier results showed that the NN bond length in the diaziridine ring is very weakly dependent on the cis- or trans-arrangement of substituents at the nitrogen atoms.
中文翻译:

6-甲基-1,5-二氮杂双环[3.1.0]己烷的平衡分子结构和光谱:气相电子衍射,量子化学和光谱数据的联合分析
通过气相电子研究了在正常条件下绝对占优势的6-甲基-1,5-二氮杂双环[3.1.0]己烷(MDABH)分子的舟构型的平衡几何形状(C s点群对称)。衍射(GED)方法在20°C下进行,涉及NMR,IR和拉曼光谱数据以及量子化学计算。在MP2 / aug-cc-pVTZ和B3LYP / cc-pVTZ水平下,计算了MDABH双环系统的环起皱变形的潜在函数。通过MP2计算发现,船形的总能量为3.52kcal mol -1低于椅子的构造。第一次,我们记录了MDABH液体样品的IR和拉曼光谱,并使用缩放量子化学力场的Pulay技术将它们的特殊性仅分配给了船形振动。在椅子形式的情况下,精制比例因子的可传递性用于可靠预测其基频的位置。根据上述数据的关节结构分析中,最重要的平衡几何ř ë被确定为用于MDABH分子的船式构象α参数(在一个键的长度;角以度为单位,Ç小号对称):C2N1 = 1.466(2),C2C3 = 1.523(2),N1N5 = 1.512(2),C6N1 = 1.440(2),C6C7 = 1.487(2),C2N1N5 = 106.1(2),N1C2C3)= 110.2(4),C2C3C4 = 99.9(4),N1N5C6 = 58.3(1),N1C6N5 = 63.3(1),N1C6C7 = 114.9(6),C6N1C2 = 111.8(1),N5N1C2C3 = 17.3( 1),N1C2C3C4 = -26.8(2),θ = C2C3C4 / C2N1N5C4 = -26.2(3),φ = N1C6N5 / C2N1N5C4 = 74.0(1)。这些结果与早期结果的比较表明,二氮丙啶环中的NN键长度非常弱地取决于氮原子上取代基的顺式或反式排列。
更新日期:2020-10-16
中文翻译:
6-甲基-1,5-二氮杂双环[3.1.0]己烷的平衡分子结构和光谱:气相电子衍射,量子化学和光谱数据的联合分析
通过气相电子研究了在正常条件下绝对占优势的6-甲基-1,5-二氮杂双环[3.1.0]己烷(MDABH)分子的舟构型的平衡几何形状(C s点群对称)。衍射(GED)方法在20°C下进行,涉及NMR,IR和拉曼光谱数据以及量子化学计算。在MP2 / aug-cc-pVTZ和B3LYP / cc-pVTZ水平下,计算了MDABH双环系统的环起皱变形的潜在函数。通过MP2计算发现,船形的总能量为3.52kcal mol -1低于椅子的构造。第一次,我们记录了MDABH液体样品的IR和拉曼光谱,并使用缩放量子化学力场的Pulay技术将它们的特殊性仅分配给了船形振动。在椅子形式的情况下,精制比例因子的可传递性用于可靠预测其基频的位置。根据上述数据的关节结构分析中,最重要的平衡几何ř ë被确定为用于MDABH分子的船式构象α参数(在一个键的长度;角以度为单位,Ç小号对称):C2N1 = 1.466(2),C2C3 = 1.523(2),N1N5 = 1.512(2),C6N1 = 1.440(2),C6C7 = 1.487(2),C2N1N5 = 106.1(2),N1C2C3)= 110.2(4),C2C3C4 = 99.9(4),N1N5C6 = 58.3(1),N1C6N5 = 63.3(1),N1C6C7 = 114.9(6),C6N1C2 = 111.8(1),N5N1C2C3 = 17.3( 1),N1C2C3C4 = -26.8(2),θ = C2C3C4 / C2N1N5C4 = -26.2(3),φ = N1C6N5 / C2N1N5C4 = 74.0(1)。这些结果与早期结果的比较表明,二氮丙啶环中的NN键长度非常弱地取决于氮原子上取代基的顺式或反式排列。










































 京公网安备 11010802027423号
京公网安备 11010802027423号