当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Accurate predictions of aqueous solubility of drug molecules via the multilevel graph convolutional network (MGCN) and SchNet architectures
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2020-09-30 , DOI: 10.1039/d0cp03596c Peng Gao 1, 2, 3, 4 , Jie Zhang 5, 6, 7, 8, 9 , Yuzhu Sun 8, 9, 10, 11 , Jianguo Yu 8, 9, 10, 11
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2020-09-30 , DOI: 10.1039/d0cp03596c Peng Gao 1, 2, 3, 4 , Jie Zhang 5, 6, 7, 8, 9 , Yuzhu Sun 8, 9, 10, 11 , Jianguo Yu 8, 9, 10, 11
Affiliation
|
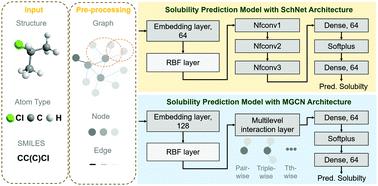
Deep learning based methods have been widely applied to predict various kinds of molecular properties in the pharmaceutical industry with increasingly more success. In this study, we propose two novel models for aqueous solubility predictions, based on the Multilevel Graph Convolutional Network (MGCN) and SchNet architectures, respectively. The advantage of the MGCN lies in the fact that it could extract the graph features of the target molecules directly from the (3D) structural information; therefore, it doesn't need to rely on a lot of intra-molecular descriptors to learn the features, which are of significance for accurate predictions of the molecular properties. The SchNet performs well in modelling the interatomic interactions inside a molecule, and such a deep learning architecture is also capable of extracting structural information and further predicting the related properties. The actual accuracy of these two novel approaches was systematically benchmarked with four different independent datasets. We found that both the MGCN and SchNet models performed well for aqueous solubility predictions. In the future, we believe such promising predictive models will be applicable to enhancing the efficiency of the screening, crystallization and delivery of drug molecules, essentially as a useful tool to promote the development of molecular pharmaceutics.
中文翻译:

通过多级图卷积网络(MGCN)和SchNet架构准确预测药物分子的水溶性
基于深度学习的方法已广泛应用于预测制药行业中各种分子特性,并且取得了越来越多的成功。在这项研究中,我们分别基于多级图卷积网络(MGCN)和SchNet体系结构,提出了两个新的水溶性预测模型。MGCN的优点在于它可以直接从(3D)结构信息中提取目标分子的图形特征;因此,它不需要依赖大量的分子内描述子来学习特征,这对于准确预测分子特性具有重要意义。SchNet在建模分子内部的原子间相互作用方面表现出色,而且这种深度学习架构还能够提取结构信息并进一步预测相关属性。这两种新颖方法的实际准确性已通过四个不同的独立数据集系统地进行了基准测试。我们发现MGCN和SchNet模型在预测水溶性方面均表现良好。将来,我们相信,这种有前途的预测模型将可用于提高药物分子的筛选,结晶和传递效率,基本上可以作为促进分子药物学发展的有用工具。我们发现MGCN和SchNet模型在预测水溶性方面均表现良好。将来,我们相信,这种有前途的预测模型将可用于提高药物分子的筛选,结晶和传递效率,基本上可以作为促进分子药物学发展的有用工具。我们发现MGCN和SchNet模型在预测水溶性方面均表现良好。将来,我们相信,这种有前途的预测模型将可用于提高药物分子的筛选,结晶和传递效率,基本上可以作为促进分子药物学发展的有用工具。
更新日期:2020-10-17
中文翻译:
通过多级图卷积网络(MGCN)和SchNet架构准确预测药物分子的水溶性
基于深度学习的方法已广泛应用于预测制药行业中各种分子特性,并且取得了越来越多的成功。在这项研究中,我们分别基于多级图卷积网络(MGCN)和SchNet体系结构,提出了两个新的水溶性预测模型。MGCN的优点在于它可以直接从(3D)结构信息中提取目标分子的图形特征;因此,它不需要依赖大量的分子内描述子来学习特征,这对于准确预测分子特性具有重要意义。SchNet在建模分子内部的原子间相互作用方面表现出色,而且这种深度学习架构还能够提取结构信息并进一步预测相关属性。这两种新颖方法的实际准确性已通过四个不同的独立数据集系统地进行了基准测试。我们发现MGCN和SchNet模型在预测水溶性方面均表现良好。将来,我们相信,这种有前途的预测模型将可用于提高药物分子的筛选,结晶和传递效率,基本上可以作为促进分子药物学发展的有用工具。我们发现MGCN和SchNet模型在预测水溶性方面均表现良好。将来,我们相信,这种有前途的预测模型将可用于提高药物分子的筛选,结晶和传递效率,基本上可以作为促进分子药物学发展的有用工具。我们发现MGCN和SchNet模型在预测水溶性方面均表现良好。将来,我们相信,这种有前途的预测模型将可用于提高药物分子的筛选,结晶和传递效率,基本上可以作为促进分子药物学发展的有用工具。










































 京公网安备 11010802027423号
京公网安备 11010802027423号