当前位置:
X-MOL 学术
›
J. Am. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Mixed-Valent Diiron µ-Carbyne, µ-Hydride Complexes: Implications for Nitrogenase
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2020-09-25 , DOI: 10.1021/jacs.0c05920 Charles H. Arnett 1 , Isabel Bogacz 2 , Ruchira Chatterjee 2 , Junko Yano 2 , Paul H. Oyala 1 , Theodor Agapie 1
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2020-09-25 , DOI: 10.1021/jacs.0c05920 Charles H. Arnett 1 , Isabel Bogacz 2 , Ruchira Chatterjee 2 , Junko Yano 2 , Paul H. Oyala 1 , Theodor Agapie 1
Affiliation
|
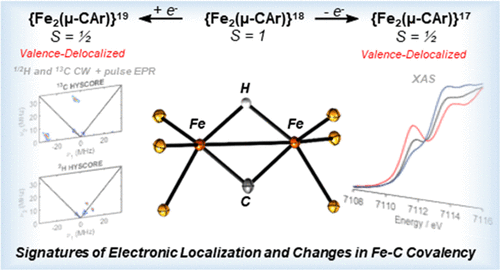
Binding of N2 by the FeMo-cofactor of nitrogenase is believed to occur after transfer of 4 e- and 4 H+ equivalents to the active site. Although pulse EPR studies indicate the presence of two Fe-(µ-H)-Fe moieties, the structural and electronic features of this mixed valent intermediate remain poorly understood. Toward an improved understanding of this bioor-ganometallic cluster, we report herein the synthesis and characterization of the first carbon-bridged, bimetallic complex-es featuring odd numbers of valence electrons, namely the diiron µ-carbyne species [(P6ArC)Fe2(µ-H)]+1 and [(P6ArC)Fe2(µ-H)]-1. Although both species are mixed valent and populate S = ½ states at low temperatures, spectro-scopic and computational studies reveal that only the cationic complex [(P6ArC)Fe2(µ-H)]1+ is valence localized, allowing an evaluation of the effects of geometrical distortions and vibronic trapping on the electronic structure of the biologically relevant Fe(µ-CAr)(µ-H)Fe motif. The influence of valence localization on the spectroscopic signature of the µ-hydride ligand was determined by 1H pulse EPR studies of [(P6ArC)Fe2(µ-H)]1+ which, compared to analogous data for the {Fe2(µ-H)}2 state of FeMoco (E4(4H)), suggests that the hydride ligands in E4(4H) bridge isovalent (most probably FeIII) metal centers. Although electron transfer involves metal-localized orbitals, investigations of [(P6ArC)Fe2(µ-H)]+1 and [(P6ArC)Fe2(µ-H)]-1 by 13C pulse EPR revealed that redox chemistry induces significant changes in Fe-C covalency (-21% upon 2 e- reduction), a conclusion further supported by X-ray absorption spectroscopy, 57Fe Mössbauer studies, and DFT calculations. Combined, our studies demonstrate that changes in covalency buffer against the accumulation of excess charge density on the metals by redistributing it to the bridging carbon, thereby facilitating multi-electron trans-formations.
中文翻译:

Mixed-Valent Diiron µ-Carbyne, µ-Hydride complexes: 对氮酶的影响
据信,固氮酶的 FeMo 辅因子与 N2 的结合发生在将 4 e- 和 4 H+ 等价物转移到活性位点后。尽管脉冲 EPR 研究表明存在两个 Fe-(µ-H)-Fe 部分,但这种混合价中间体的结构和电子特征仍然知之甚少。为了更好地理解这种生物有机金属簇,我们在此报告了第一个具有奇数价电子的碳桥联双金属配合物的合成和表征,即二铁 μ-碳炔物质 [(P6ArC)Fe2(μ -H)]+1 和 [(P6ArC)Fe2(µ-H)]-1。尽管这两种物质都是混合价态并在低温下填充 S = ½ 态,但光谱和计算研究表明,只有阳离子络合物 [(P6ArC)Fe2(µ-H)]1+ 是价定域的,允许评估几何变形和振动捕获对生物相关 Fe(µ-CAr)(µ-H)Fe 基序的电子结构的影响。价定位对 μ-氢化物配体光谱特征的影响由 [(P6ArC)Fe2(μ-H)]1+ 的 1H 脉冲 EPR 研究确定,与 {Fe2(μ-H) 的类似数据相比)}FeMoco (E4(4H)) 的 2 态,表明 E4(4H) 中的氢化物配体桥接等价(最可能是 FeIII)金属中心。尽管电子转移涉及金属定域轨道,但通过 13C 脉冲 EPR 对 [(P6ArC)Fe2(µ-H)]+1 和 [(P6ArC)Fe2(µ-H)]-1 的研究表明,氧化还原化学导致Fe-C 共价(2 e 还原时为 -21%),这一结论得到 X 射线吸收光谱、57Fe Mössbauer 研究和 DFT 计算的进一步支持。
更新日期:2020-09-25
中文翻译:
Mixed-Valent Diiron µ-Carbyne, µ-Hydride complexes: 对氮酶的影响
据信,固氮酶的 FeMo 辅因子与 N2 的结合发生在将 4 e- 和 4 H+ 等价物转移到活性位点后。尽管脉冲 EPR 研究表明存在两个 Fe-(µ-H)-Fe 部分,但这种混合价中间体的结构和电子特征仍然知之甚少。为了更好地理解这种生物有机金属簇,我们在此报告了第一个具有奇数价电子的碳桥联双金属配合物的合成和表征,即二铁 μ-碳炔物质 [(P6ArC)Fe2(μ -H)]+1 和 [(P6ArC)Fe2(µ-H)]-1。尽管这两种物质都是混合价态并在低温下填充 S = ½ 态,但光谱和计算研究表明,只有阳离子络合物 [(P6ArC)Fe2(µ-H)]1+ 是价定域的,允许评估几何变形和振动捕获对生物相关 Fe(µ-CAr)(µ-H)Fe 基序的电子结构的影响。价定位对 μ-氢化物配体光谱特征的影响由 [(P6ArC)Fe2(μ-H)]1+ 的 1H 脉冲 EPR 研究确定,与 {Fe2(μ-H) 的类似数据相比)}FeMoco (E4(4H)) 的 2 态,表明 E4(4H) 中的氢化物配体桥接等价(最可能是 FeIII)金属中心。尽管电子转移涉及金属定域轨道,但通过 13C 脉冲 EPR 对 [(P6ArC)Fe2(µ-H)]+1 和 [(P6ArC)Fe2(µ-H)]-1 的研究表明,氧化还原化学导致Fe-C 共价(2 e 还原时为 -21%),这一结论得到 X 射线吸收光谱、57Fe Mössbauer 研究和 DFT 计算的进一步支持。










































 京公网安备 11010802027423号
京公网安备 11010802027423号