Chemical Physics Letters ( IF 2.8 ) Pub Date : 2020-09-24 , DOI: 10.1016/j.cplett.2020.138026 Chongchong She , Kun Chen , Shaohua Jin , Lijie Li , Shusen Chen , Huanmin Liu , Wei Liu , Fang Bao
|
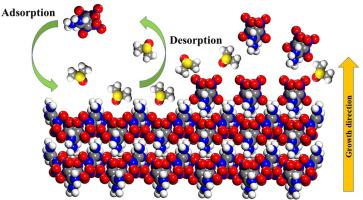
A computational strategy in consideration of the crystal surface structures was proposed based on the attachment energy model to unravel of the effect of solvent on the crystal morphology. The proposed strategy is confirmed by its successful applications to predict the crystal morphology of FOX-7 growth in DMSO. The analysis results of the binding energy composition between the solvent and the crystal show that the adsorption of DMSO molecules on the surface with nitro groups is mainly through forming hydrogen bonds, whereas the adsorption on the surface with amino groups is mainly due to the van der Waals interaction.
中文翻译:
考虑晶体表面结构的高能材料晶体形态预测计算策略
基于附着能模型,提出了考虑晶体表面结构的计算策略,以揭示溶剂对晶体形态的影响。所提出的策略已被成功应用于预测DMSO中FOX-7生长的晶体形态而得到证实。溶剂与晶体之间结合能组成的分析结果表明,DMSO分子在硝基表面的吸附主要是通过形成氢键,而在氨基表面的吸附主要是由于范德华尔互动。










































 京公网安备 11010802027423号
京公网安备 11010802027423号