当前位置:
X-MOL 学术
›
Adv. Theory Simul.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Accelerated Modeling of Lithium Diffusion in Solid State Electrolytes using Artificial Neural Networks
Advanced Theory and Simulations ( IF 2.9 ) Pub Date : 2020-07-16 , DOI: 10.1002/adts.202000097 Karun K. Rao 1, 2 , Yan Yao 2, 3 , Lars C. Grabow 1, 2
Advanced Theory and Simulations ( IF 2.9 ) Pub Date : 2020-07-16 , DOI: 10.1002/adts.202000097 Karun K. Rao 1, 2 , Yan Yao 2, 3 , Lars C. Grabow 1, 2
Affiliation
|
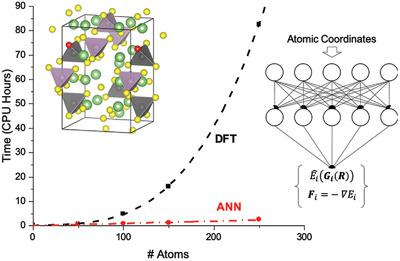
Previous efforts to understand structure‐function relationships in high ionic conductivity materials for solid state batteries have predominantly relied on density functional theory (DFT‐) based ab initio molecular dynamics (MD). Such simulations, however, are computationally demanding and cannot be reasonably applied to large systems containing more than a hundred atoms. Here, an artificial neural network (ANN) is trained to accelerate the calculation of high accuracy atomic forces and energies used during such MD simulations. After carefully training a robust ANN for four and five element systems, nearly identical lithium ion diffusivities are obtained for Li10GeP2S12 (LGPS) when benchmarking the ANN‐MD results with DFT‐MD. Applying the ANN‐MD approach, the effect of chlorine doping on the lithium diffusivity is calculated in an LGPS‐like structure and it is found that a dopant concentration of 1.3% maximizes ionic conductivity. The optimal concentration balances the competing consequences of effective atomic radii and dielectric constants on lithium diffusion and agrees with the experimental composition. Performing simulations at the resolution necessary to model experimentally relevant and optimal concentrations would be infeasible with traditional DFT‐MD. Systems that require a large number of simulated atoms can be studied more efficiently while maintaining high accuracy with the proposed ANN‐MD framework.
中文翻译:

使用人工神经网络加速锂在固态电解质中的扩散建模
以前了解固态电池高离子电导率材料中结构-功能关系的努力主要依靠基于密度泛函理论(DFT-)的从头算分子动力学(MD)。但是,这种模拟对计算的要求很高,不能合理地应用于包含一百个以上原子的大型系统。在这里,训练了一个人工神经网络(ANN),以加速在此类MD模拟过程中使用的高精度原子力和能量的计算。在仔细训练了针对四元和五元系统的鲁棒人工神经网络之后,对于Li 10 GeP 2 S 12,获得了几乎相同的锂离子扩散率(LGPS)将DFT-MD用作基准ANN-MD结果时。应用ANN-MD方法,在类似LGPS的结构中计算了氯掺杂对锂扩散率的影响,发现1.3%的掺杂剂浓度可使离子电导率最大化。最佳浓度平衡了有效原子半径和介电常数对锂扩散的竞争结果,并且与实验组成一致。使用传统的DFT-MD,以模拟实验相关的最佳浓度所需的分辨率执行模拟将是不可行的。可以使用拟议的ANN-MD框架在保持高精度的同时更有效地研究需要大量模拟原子的系统。
更新日期:2020-09-23
中文翻译:
使用人工神经网络加速锂在固态电解质中的扩散建模
以前了解固态电池高离子电导率材料中结构-功能关系的努力主要依靠基于密度泛函理论(DFT-)的从头算分子动力学(MD)。但是,这种模拟对计算的要求很高,不能合理地应用于包含一百个以上原子的大型系统。在这里,训练了一个人工神经网络(ANN),以加速在此类MD模拟过程中使用的高精度原子力和能量的计算。在仔细训练了针对四元和五元系统的鲁棒人工神经网络之后,对于Li 10 GeP 2 S 12,获得了几乎相同的锂离子扩散率(LGPS)将DFT-MD用作基准ANN-MD结果时。应用ANN-MD方法,在类似LGPS的结构中计算了氯掺杂对锂扩散率的影响,发现1.3%的掺杂剂浓度可使离子电导率最大化。最佳浓度平衡了有效原子半径和介电常数对锂扩散的竞争结果,并且与实验组成一致。使用传统的DFT-MD,以模拟实验相关的最佳浓度所需的分辨率执行模拟将是不可行的。可以使用拟议的ANN-MD框架在保持高精度的同时更有效地研究需要大量模拟原子的系统。










































 京公网安备 11010802027423号
京公网安备 11010802027423号