当前位置:
X-MOL 学术
›
Adv. Theory Simul.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
A First‐Principles‐Based Sub‐Lattice Formalism for Predicting Off‐Stoichiometry in Materials for Solar Thermochemical Applications: The Example of Ceria
Advanced Theory and Simulations ( IF 2.9 ) Pub Date : 2020-08-17 , DOI: 10.1002/adts.202000112 Gopalakrishnan Sai Gautam 1 , Ellen B. Stechel 2 , Emily A. Carter 1, 3
Advanced Theory and Simulations ( IF 2.9 ) Pub Date : 2020-08-17 , DOI: 10.1002/adts.202000112 Gopalakrishnan Sai Gautam 1 , Ellen B. Stechel 2 , Emily A. Carter 1, 3
Affiliation
|
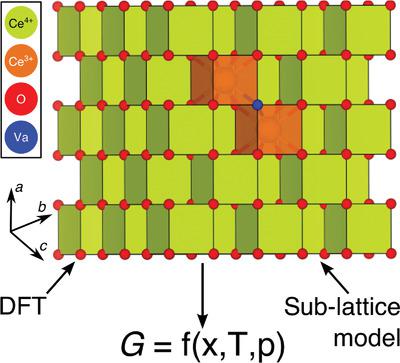
Theoretical models that reliably can predict off‐stoichiometry in materials via accurate descriptions of underlying thermodynamics are crucial for energy applications. For example, transition‐metal and rare‐earth oxides that can tolerate a large number of oxygen vacancies, such as CeO2 and doped CeO2, can split water and carbon dioxide via a two‐step, oxide‐based solar thermochemical (STC) cycle. The search for new STC materials with a performance superior to that of state‐of‐the‐art CeO2 can benefit from predictions accurately describing the thermodynamics of oxygen vacancies. The sub‐lattice formalism, a common tool used to fit experimental data and build temperature‐composition phase diagrams, can be useful in this context. Here, sub‐lattice models are derived solely from zero‐temperature quantum mechanics calculations to estimate fairly accurate temperature‐ and oxygen‐partial‐pressure‐dependent off‐stoichiometries in CeO2 and Zr‐doped CeO2. Physical motivations for deriving some of the “excess” sub‐lattice model parameters directly from quantum mechanical calculations, instead of fitting to minimize deviations from experimental and/or theoretical data, are identified. Important limitations and approximations of the approach used are specified and extensions to multi‐cation oxides are also suggested to help identify novel candidates for water and carbon dioxide splitting and related applications.
中文翻译:

基于第一原理的子格形式主义,用于预测太阳热化学应用材料的化学计量失误:Ceria的示例
通过对基本热力学的准确描述,可以可靠地预测材料化学计量失误的理论模型对于能源应用至关重要。例如,可以容忍大量氧空位的过渡金属和稀土氧化物,例如CeO 2和掺杂的CeO 2,可以通过基于氧化物的两步式太阳能热化学(STC)分解水和二氧化碳周期。寻找性能优于最新CeO 2的新STC材料可以得益于准确描述氧空位热力学的预测。在这种情况下,子晶格形式主义是用于拟合实验数据和建立温度组成相图的常用工具。在这里,亚晶格模型仅从零温度量子力学计算中得出,以估算CeO 2和Zr掺杂CeO 2中相当精确的温度和氧分压依赖性非化学计量比。。确定了直接从量子力学计算中导出“多余的”亚晶格模型参数的物理动机,而不是为了使与实验和/或理论数据的偏差最小化而进行拟合。详细说明了所用方法的重要局限性和近似性,并建议对多阳离子氧化物进行扩展,以帮助确定水和二氧化碳分解及相关应用的新候选物。
更新日期:2020-09-23
中文翻译:
基于第一原理的子格形式主义,用于预测太阳热化学应用材料的化学计量失误:Ceria的示例
通过对基本热力学的准确描述,可以可靠地预测材料化学计量失误的理论模型对于能源应用至关重要。例如,可以容忍大量氧空位的过渡金属和稀土氧化物,例如CeO 2和掺杂的CeO 2,可以通过基于氧化物的两步式太阳能热化学(STC)分解水和二氧化碳周期。寻找性能优于最新CeO 2的新STC材料可以得益于准确描述氧空位热力学的预测。在这种情况下,子晶格形式主义是用于拟合实验数据和建立温度组成相图的常用工具。在这里,亚晶格模型仅从零温度量子力学计算中得出,以估算CeO 2和Zr掺杂CeO 2中相当精确的温度和氧分压依赖性非化学计量比。。确定了直接从量子力学计算中导出“多余的”亚晶格模型参数的物理动机,而不是为了使与实验和/或理论数据的偏差最小化而进行拟合。详细说明了所用方法的重要局限性和近似性,并建议对多阳离子氧化物进行扩展,以帮助确定水和二氧化碳分解及相关应用的新候选物。










































 京公网安备 11010802027423号
京公网安备 11010802027423号