当前位置:
X-MOL 学术
›
Phys. Rev. B
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Density functional and classical simulations of liquid and glassy selenium
Physical Review B ( IF 3.2 ) Pub Date : 2020-09-22 , DOI: 10.1103/physrevb.102.104202 J. Kalikka , J. Akola , R. O. Jones , H. R. Schober
Physical Review B ( IF 3.2 ) Pub Date : 2020-09-22 , DOI: 10.1103/physrevb.102.104202 J. Kalikka , J. Akola , R. O. Jones , H. R. Schober
|
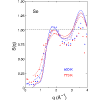
Molecular dynamics simulations of liquid and glassy selenium have been carried out using density functional (400–773 K, 600 atoms) and classical force field (290–500 K, 5488 atoms) methods. Structural features (structure factors, pair distribution functions, bond lengths, bond and dihedral angles, cavities) and dynamical properties (diffusion coefficients, power spectra, sound velocity, collective excitations, bond lifetimes) agree well with experimental data where available. The structures are predominantly chainlike, with a small fraction of rings with a range of sizes, and large cavity volumes lead to flexible chains. It is striking that the density functional simulations show very few rings at 600 K and below.
中文翻译:

液态和玻璃态硒的密度泛函和经典模拟
使用密度泛函(400–773 K,600原子)和经典力场(290–500 K,5488原子)方法进行了液态和玻璃态硒的分子动力学模拟。结构特征(结构因子,线对分布函数,键长,键和二面角,腔)和动力学特性(扩散系数,功率谱,声速,集体激发,键寿命)与实验数据一致。该结构主要是链状的,有一小部分具有一定大小范围的环,并且大的空腔体积会导致柔性链。令人惊讶的是,密度泛函模拟显示很少 在600 K及以下时响铃。
更新日期:2020-09-22
中文翻译:
液态和玻璃态硒的密度泛函和经典模拟
使用密度泛函(400–773 K,600原子)和经典力场(290–500 K,5488原子)方法进行了液态和玻璃态硒的分子动力学模拟。结构特征(结构因子,线对分布函数,键长,键和二面角,腔)和动力学特性(扩散系数,功率谱,声速,集体激发,键寿命)与实验数据一致。该结构主要是链状的,有一小部分具有一定大小范围的环,并且大的空腔体积会导致柔性链。令人惊讶的是,密度泛函模拟显示很少 在600 K及以下时响铃。










































 京公网安备 11010802027423号
京公网安备 11010802027423号