当前位置:
X-MOL 学术
›
Surf. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
First-principles investigation of phosphate ester and carboxylic acid on BaTiO3 surfaces with stoichiometric terminations
Surface Science ( IF 1.9 ) Pub Date : 2021-01-01 , DOI: 10.1016/j.susc.2020.121737 Hee-Joon Chun , Hyung Kyu Kim , Younghwan Yoon , Seong-Chan Park
Surface Science ( IF 1.9 ) Pub Date : 2021-01-01 , DOI: 10.1016/j.susc.2020.121737 Hee-Joon Chun , Hyung Kyu Kim , Younghwan Yoon , Seong-Chan Park
|
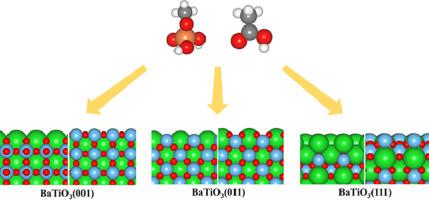
Abstract Phosphate ester and carboxylic acid are common anchoring groups for organic dispersants which are used in BaTiO3 suspension processes for multi-layered ceramic capacitors (MLCCs). However, the adsorption behaviors and the structure sensitivity of phosphate ester and carboxylic acid on BaTiO3 surfaces at the molecular-level remain largely unknown and thus require a systematic study. In this report, we theoretically investigate adsorption of those molecules on the BaTiO3 surfaces using Density Functional Theory (DFT). Our study includes three low-index facets of BaTiO3: the BaTiO3(001), (011), and (111) surfaces with the stoichiometric terminations. Also, to model phosphate ester and carboxylic acid, we employ methyl dihydrogen phosphate (CH3OPO3H2) and acetic acid (CH3COOH) in our simulation. Our DFT result reveals that BaTiO3(011)-BaTiO is the most active surface for molecular and dissociative adsorption of phosphate ester and carboxylic acid compared to other surfaces. Also, the saturated coverages of phosphate ester and carboxylic acid on the BaTiO3(011)-BaTiO surface are 0.5 ML (monolayer), respectively, based on the grand potential analysis. These findings reveal the role of the BaTiO3 facets and terminations for adsorption of phosphate ester and carboxylic acid. Thus, we believe that our DFT result will be a building block for understanding on the interaction of tetragonal BaTiO3 with relevant dispersants which are utilized in MLCC applications.
中文翻译:

具有化学计量末端的 BaTiO3 表面磷酸酯和羧酸的第一性原理研究
摘要 磷酸酯和羧酸是有机分散剂的常见锚定基团,用于多层陶瓷电容器 (MLCC) 的 BaTiO3 悬浮工艺。然而,磷酸酯和羧酸在分子水平上对 BaTiO3 表面的吸附行为和结构敏感性仍然很大程度上未知,因此需要系统研究。在本报告中,我们使用密度泛函理论 (DFT) 从理论上研究了这些分子在 BaTiO3 表面上的吸附。我们的研究包括 BaTiO3 的三个低指数面:具有化学计量终止的 BaTiO3(001)、(011) 和 (111) 表面。此外,为了模拟磷酸酯和羧酸,我们在模拟中使用了磷酸二氢甲酯 (CH3OPO3H2) 和乙酸 (CH3COOH)。我们的 DFT 结果表明,与其他表面相比,BaTiO3(011)-BaTiO 是磷酸酯和羧酸的分子和解离吸附最活跃的表面。此外,基于大电位分析,磷酸酯和羧酸在 BaTiO3(011)-BaTiO 表面的饱和覆盖率分别为 0.5 ML(单层)。这些发现揭示了 BaTiO3 小平面和末端对磷酸酯和羧酸吸附的作用。因此,我们相信我们的 DFT 结果将成为理解四方 BaTiO3 与 MLCC 应用中使用的相关分散剂相互作用的基础。根据大电位分析,磷酸酯和羧酸在 BaTiO3(011)-BaTiO 表面的饱和覆盖率分别为 0.5 ML(单层)。这些发现揭示了 BaTiO3 小平面和末端对磷酸酯和羧酸吸附的作用。因此,我们相信我们的 DFT 结果将成为理解四方 BaTiO3 与 MLCC 应用中使用的相关分散剂相互作用的基础。根据大电位分析,磷酸酯和羧酸在 BaTiO3(011)-BaTiO 表面的饱和覆盖率分别为 0.5 ML(单层)。这些发现揭示了 BaTiO3 小平面和末端对磷酸酯和羧酸吸附的作用。因此,我们相信我们的 DFT 结果将成为理解四方 BaTiO3 与 MLCC 应用中使用的相关分散剂相互作用的基础。
更新日期:2021-01-01
中文翻译:
具有化学计量末端的 BaTiO3 表面磷酸酯和羧酸的第一性原理研究
摘要 磷酸酯和羧酸是有机分散剂的常见锚定基团,用于多层陶瓷电容器 (MLCC) 的 BaTiO3 悬浮工艺。然而,磷酸酯和羧酸在分子水平上对 BaTiO3 表面的吸附行为和结构敏感性仍然很大程度上未知,因此需要系统研究。在本报告中,我们使用密度泛函理论 (DFT) 从理论上研究了这些分子在 BaTiO3 表面上的吸附。我们的研究包括 BaTiO3 的三个低指数面:具有化学计量终止的 BaTiO3(001)、(011) 和 (111) 表面。此外,为了模拟磷酸酯和羧酸,我们在模拟中使用了磷酸二氢甲酯 (CH3OPO3H2) 和乙酸 (CH3COOH)。我们的 DFT 结果表明,与其他表面相比,BaTiO3(011)-BaTiO 是磷酸酯和羧酸的分子和解离吸附最活跃的表面。此外,基于大电位分析,磷酸酯和羧酸在 BaTiO3(011)-BaTiO 表面的饱和覆盖率分别为 0.5 ML(单层)。这些发现揭示了 BaTiO3 小平面和末端对磷酸酯和羧酸吸附的作用。因此,我们相信我们的 DFT 结果将成为理解四方 BaTiO3 与 MLCC 应用中使用的相关分散剂相互作用的基础。根据大电位分析,磷酸酯和羧酸在 BaTiO3(011)-BaTiO 表面的饱和覆盖率分别为 0.5 ML(单层)。这些发现揭示了 BaTiO3 小平面和末端对磷酸酯和羧酸吸附的作用。因此,我们相信我们的 DFT 结果将成为理解四方 BaTiO3 与 MLCC 应用中使用的相关分散剂相互作用的基础。根据大电位分析,磷酸酯和羧酸在 BaTiO3(011)-BaTiO 表面的饱和覆盖率分别为 0.5 ML(单层)。这些发现揭示了 BaTiO3 小平面和末端对磷酸酯和羧酸吸附的作用。因此,我们相信我们的 DFT 结果将成为理解四方 BaTiO3 与 MLCC 应用中使用的相关分散剂相互作用的基础。


























 京公网安备 11010802027423号
京公网安备 11010802027423号