当前位置:
X-MOL 学术
›
Acta Crystallogr. A Found. Adv.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Fast analytical evaluation of intermolecular electrostatic interaction energies using the pseudoatom representation of the electron density. III. Application to crystal structures via the Ewald and direct summation methods
Acta Crystallographica Section A: Foundations and Advances ( IF 1.9 ) Pub Date : 2020-09-18 , DOI: 10.1107/s2053273320009584 Daniel Nguyen , Piero Macchi , Anatoliy Volkov
Acta Crystallographica Section A: Foundations and Advances ( IF 1.9 ) Pub Date : 2020-09-18 , DOI: 10.1107/s2053273320009584 Daniel Nguyen , Piero Macchi , Anatoliy Volkov
|
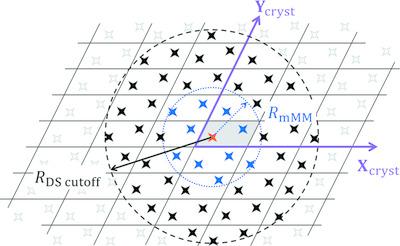
The previously reported exact potential and multipole moment (EP/MM) method for fast and accurate evaluation of the intermolecular electrostatic interaction energies using the pseudoatom representation of the electron density [Volkov, Koritsanszky & Coppens (2004). Chem. Phys. Lett.391, 170–175; Nguyen, Kisiel & Volkov (2018). Acta Cryst. A74, 524–536; Nguyen & Volkov (2019). Acta Cryst. A75, 448–464] is extended to the calculation of electrostatic interaction energies in molecular crystals using two newly developed implementations: (i) the Ewald summation (ES), which includes interactions up to the hexadecapolar level and the EP correction to account for short‐range electron‐density penetration effects, and (ii) the enhanced EP/MM‐based direct summation (DS), which at sufficiently large intermolecular separations replaces the atomic multipole moment approximation to the electrostatic energy with that based on the molecular multipole moments. As in the previous study [Nguyen, Kisiel & Volkov (2018). Acta Cryst. A74, 524–536], the EP electron repulsion integral is evaluated analytically using the Löwdin α‐function approach. The resulting techniques, incorporated in the XDPROP module of the software package XD2016, have been tested on several small‐molecule crystal systems (benzene, l‐dopa, paracetamol, amino acids etc.) and the crystal structure of a 181‐atom decapeptide molecule (Z = 4) using electron densities constructed via the University at Buffalo Aspherical Pseudoatom Databank [Volkov, Li, Koritsanszky & Coppens (2004). J. Phys. Chem. A, 108, 4283–4300]. Using a 2015 2.8 GHz Intel Xeon E3‐1505M v5 computer processor, a 64‐bit implementation of the Löwdin α‐function and one of the higher optimization levels in the GNU Fortran compiler, the ES method evaluates the electrostatic interaction energy with a numerical precision of at least 10−5 kJ mol−1 in under 6 s for any of the tested small‐molecule crystal structures, and in 48.5 s for the decapeptide structure. The DS approach is competitive in terms of precision and speed with the ES technique only for crystal structures of small molecules that do not carry a large molecular dipole moment. The electron‐density penetration effects, correctly accounted for by the two described methods, contribute 28–64% to the total electrostatic interaction energy in the examined systems, and thus cannot be neglected.
中文翻译:

使用电子密度的假原子表示,快速分析评估分子间静电相互作用能。三,通过Ewald和直接求和方法应用于晶体结构
先前报道的精确电势和多极矩(EP / MM)方法使用电子密度的伪原子表示来快速而准确地评估分子间静电相互作用能[Volkov,Koritsanszky&Coppens(2004)。化学 物理 来吧 391,170-175; 阮基西尔和沃尔科夫(2018)。Acta Cryst。甲74,524-536; Nguyen&Volkov(2019)。Acta Cryst。A 75,448–464]扩展到使用两个新开发的实现方法来计算分子晶体中的静电相互作用能:(i)埃瓦尔德求和(ES),包括高达十六极级的相互作用和EP校正,以解决短时范围的电子密度穿透效应,以及(ii)增强的基于EP / MM的直接求和(DS),在足够大的分子间间距下,原子多极矩近似值取代了基于分子多极矩的静电能。与之前的研究一样[Nguyen,Kisiel&Volkov(2018)。Acta Cryst。甲74,524-536],在EP电子斥力积分解析使用Löwdinα-函数方法进行评价。由此产生的技术,并入XDPROP软件包的模块XD2016,已经对几个小分子晶体系统(苯,测试升-DOPA,扑热息痛,氨基酸等)和181-原子十肽分子(晶体结构Ž = 4),使用电子通过布法罗非球面伪原子数据库在大学中构建的密度[Volkov,Li,Koritsanszky&Coppens(2004)。J.物理 化学 A,108,4283–4300]。ES方法使用2015年的2.8 GHz Intel Xeon E3‐1505M v5计算机处理器,Löwdinα函数的64位实现以及GNU Fortran编译器中的更高优化级别之一,可以以数值精度评估静电相互作用能。至少10 -5 kJ mol -1对于任何测试的小分子晶体结构,在6 s内即可完成;对于十肽结构,则在48.5 s内即可完成。DS方法仅在不携带大分子偶极矩的小分子晶体结构中,就ES技术而言在精度和速度方面具有竞争力。两种描述方法正确解释的电子密度渗透效应,在所检查的系统中占总静电相互作用能的28-64%,因此不能忽略。
更新日期:2020-11-02
中文翻译:
使用电子密度的假原子表示,快速分析评估分子间静电相互作用能。三,通过Ewald和直接求和方法应用于晶体结构
先前报道的精确电势和多极矩(EP / MM)方法使用电子密度的伪原子表示来快速而准确地评估分子间静电相互作用能[Volkov,Koritsanszky&Coppens(2004)。化学 物理 来吧 391,170-175; 阮基西尔和沃尔科夫(2018)。Acta Cryst。甲74,524-536; Nguyen&Volkov(2019)。Acta Cryst。A 75,448–464]扩展到使用两个新开发的实现方法来计算分子晶体中的静电相互作用能:(i)埃瓦尔德求和(ES),包括高达十六极级的相互作用和EP校正,以解决短时范围的电子密度穿透效应,以及(ii)增强的基于EP / MM的直接求和(DS),在足够大的分子间间距下,原子多极矩近似值取代了基于分子多极矩的静电能。与之前的研究一样[Nguyen,Kisiel&Volkov(2018)。Acta Cryst。甲74,524-536],在EP电子斥力积分解析使用Löwdinα-函数方法进行评价。由此产生的技术,并入XDPROP软件包的模块XD2016,已经对几个小分子晶体系统(苯,测试升-DOPA,扑热息痛,氨基酸等)和181-原子十肽分子(晶体结构Ž = 4),使用电子通过布法罗非球面伪原子数据库在大学中构建的密度[Volkov,Li,Koritsanszky&Coppens(2004)。J.物理 化学 A,108,4283–4300]。ES方法使用2015年的2.8 GHz Intel Xeon E3‐1505M v5计算机处理器,Löwdinα函数的64位实现以及GNU Fortran编译器中的更高优化级别之一,可以以数值精度评估静电相互作用能。至少10 -5 kJ mol -1对于任何测试的小分子晶体结构,在6 s内即可完成;对于十肽结构,则在48.5 s内即可完成。DS方法仅在不携带大分子偶极矩的小分子晶体结构中,就ES技术而言在精度和速度方面具有竞争力。两种描述方法正确解释的电子密度渗透效应,在所检查的系统中占总静电相互作用能的28-64%,因此不能忽略。










































 京公网安备 11010802027423号
京公网安备 11010802027423号