当前位置:
X-MOL 学术
›
Mol. Pharmacol.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
GS-967 and Eleclazine Block Sodium Channels in Human Induced Pluripotent Stem Cell-derived Cardiomyocytes.
Molecular Pharmacology ( IF 3.2 ) Pub Date : 2020-11-01 , DOI: 10.1124/molpharm.120.000048 Franck Potet 1 , Defne E Egecioglu 1 , Paul W Burridge 1 , Alfred L George 2
Molecular Pharmacology ( IF 3.2 ) Pub Date : 2020-11-01 , DOI: 10.1124/molpharm.120.000048 Franck Potet 1 , Defne E Egecioglu 1 , Paul W Burridge 1 , Alfred L George 2
Affiliation
|
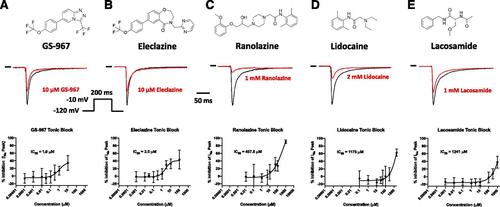
GS-967 and eleclazine (GS-6615) are novel sodium channel inhibitors exhibiting antiarrhythmic effects in various in vitro and in vivo models. The antiarrhythmic mechanism has been attributed to preferential suppression of late sodium current (INaL). Here, we took advantage of a high throughput automated electrophysiology platform (SyncroPatch 768PE) to investigate the molecular pharmacology of GS-967 and eleclazine on peak sodium current (INaP) recorded from human induced pluripotent stem cell–derived cardiomyocytes. We compared the effects of GS-967 and eleclazine with the antiarrhythmic drug lidocaine, the prototype INaL inhibitor ranolazine, and the slow inactivation enhancing drug lacosamide. In human induced pluripotent stem cell–derived cardiomyocytes, GS-967 and eleclazine caused a reduction of INaP in a frequency-dependent manner consistent with use-dependent block (UDB). GS-967 and eleclazine had similar efficacy but evoked more potent UDB of INaP (IC50 = 0.07 and 0.6 µM, respectively) than ranolazine (7.8 µM), lidocaine (133.5 µM), and lacosamide (158.5 µM). In addition, GS-967 and eleclazine exerted more potent effects on slow inactivation and recovery from inactivation compared with the other sodium channel blocking drugs we tested. The greater UDB potency of GS-967 and eleclazine was attributed to the higher association rates and moderate unbinding rate of these two compounds with sodium channels. We propose that substantial UDB contributes to the observed antiarrhythmic efficacy of GS-967 and eleclazine.
中文翻译:

GS-967和Eleclazine阻断人诱导的多能干细胞衍生的心肌细胞中的钠通道。
GS-967和eleclazine(GS-6615)是在各种体外和体内模型中均表现出抗心律失常作用的新型钠通道抑制剂。抗心律不齐的机制已归因于优先抑制晚期钠电流(I NaL)。在这里,我们利用高通量自动化电生理平台(SyncroPatch 768PE)来研究GS-967和eleclazine对从人诱导的多能干细胞衍生的心肌细胞记录的峰值钠电流(I NaP)的分子药理作用。我们将GS-967和eleclazine与抗心律不齐药物利多卡因(原型I NaL)的作用进行了比较抑制剂雷诺嗪,以及缓慢失活的增强药物拉考酰胺。在人诱导的多能干细胞衍生的心肌细胞中,GS-967和伊来那嗪以频率依赖性方式导致I NaP降低,与使用依赖性阻断(UDB)一致。GS-967和eleclazine具有相似的功效,但诱发了更强的I NaP UDB (IC 50分别为雷诺嗪(7.8 µM),利多卡因(133.5 µM)和拉可酰胺(158.5 µM)的0.07和0.6 µM。此外,与我们测试的其他钠通道阻滞药物相比,GS-967和伊曲嗪对缓慢的失活和从失活中恢复的作用更大。GS-967和eleclazine的UDB效能更高是由于这两种具有钠通道的化合物具有更高的缔合速率和中等的解键速率。我们建议大量的UDB有助于观察到的GS-967和eleclazine的抗心律不齐功效。
更新日期:2020-10-13
中文翻译:
GS-967和Eleclazine阻断人诱导的多能干细胞衍生的心肌细胞中的钠通道。
GS-967和eleclazine(GS-6615)是在各种体外和体内模型中均表现出抗心律失常作用的新型钠通道抑制剂。抗心律不齐的机制已归因于优先抑制晚期钠电流(I NaL)。在这里,我们利用高通量自动化电生理平台(SyncroPatch 768PE)来研究GS-967和eleclazine对从人诱导的多能干细胞衍生的心肌细胞记录的峰值钠电流(I NaP)的分子药理作用。我们将GS-967和eleclazine与抗心律不齐药物利多卡因(原型I NaL)的作用进行了比较抑制剂雷诺嗪,以及缓慢失活的增强药物拉考酰胺。在人诱导的多能干细胞衍生的心肌细胞中,GS-967和伊来那嗪以频率依赖性方式导致I NaP降低,与使用依赖性阻断(UDB)一致。GS-967和eleclazine具有相似的功效,但诱发了更强的I NaP UDB (IC 50分别为雷诺嗪(7.8 µM),利多卡因(133.5 µM)和拉可酰胺(158.5 µM)的0.07和0.6 µM。此外,与我们测试的其他钠通道阻滞药物相比,GS-967和伊曲嗪对缓慢的失活和从失活中恢复的作用更大。GS-967和eleclazine的UDB效能更高是由于这两种具有钠通道的化合物具有更高的缔合速率和中等的解键速率。我们建议大量的UDB有助于观察到的GS-967和eleclazine的抗心律不齐功效。










































 京公网安备 11010802027423号
京公网安备 11010802027423号