Journal of Catalysis ( IF 7.3 ) Pub Date : 2020-09-17 , DOI: 10.1016/j.jcat.2020.09.007 Takeshi Nishimoto , Tatsuya Shinagawa , Takahiro Naito , Kazuhiro Takanabe
|
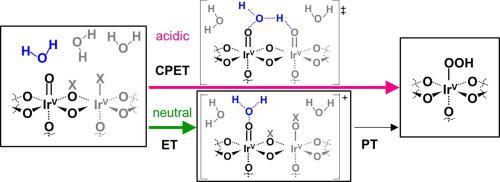
Water electrolysis driven by electrical power generated from renewable energy sources will play a pivotal role in future sustainable societies, which requires adaptation of various reaction conditions as well as electrolyte identities. Regardless, the anodic half-reaction of the oxygen evolution reaction (OER) is considered a kinetic bottleneck. This study provides quantitative description of the OER kinetics based on rigorous microkinetic analyses including Tafel analysis, isotope effects and temperature dependence using an IrOx electrocatalyst in unbuffered solution at varying pH levels. The diffusional constraints of H+/OH− determine three distinctive kinetic regimes in the pH-potential-current relationships: below pH 5, between pH 5 and 10, and above pH 10 at appreciable current densities on the order of 1 mA cm−2. When shifting from alkaline to acidic solution, the complete consumption of local OH− near the electrode surface switches the OER proceeding as the oxidation of OH− to that of the water molecule at pH ~ 11 irrespective of the electrode identity. At pH 5–10, the diffusional constraints of H+ generated via oxidation reaction yield an environment with pH ~ 4 near the electrode surface even prior to the OER, resulting in a bulk pH-independent region for the OER performance. Under this unbuffered near-neutral-pH condition, the isotope effect was diminished for the OER catalysis, which is consistent with the rate-determining step (rds) being the sole electron-transfer step via the formation of O-O bonds, decoupled from proton transfer. This reaction mechanism is distinct from that under more acidic conditions (pH < 4), although the water molecule is the same reactant. Under acidic conditions, noticeable isotope effects were observable, which is consistent with the formation of O-O bonds being the rds on uncoordinated bare Ir sites as the most abundant surface species. This study provides a quantitative description of the reactant- and mechanistic-switching that points to concurrent optimization of both electrode materials and electrolyte for improved OER performance at near-neutral pH levels.
中文翻译:
在非缓冲条件下氧化铱上电催化氧释放反应的微动力学评估
由可再生能源产生的电能驱动的水电解将在未来的可持续社会中发挥关键作用,这需要适应各种反应条件以及电解质特性。无论如何,氧气逸出反应(OER)的阳极半反应被认为是动力学瓶颈。这项研究基于严格的微动力学分析,包括Tafel分析,同位素效应和使用IrO x电催化剂在不同pH值的非缓冲溶液中对温度的依赖性,对OER动力学进行了定量描述。H的扩散限制+ / OH -确定pH电位-电流关系中的三种独特的动力学机制:pH低于5,pH介于5和10之间以及pH高于10处于明显的电流密度(约为1 mA cm -2)。当从碱性转移到酸性溶液中,本地OH的完全消耗-在电极表面附近切换OER程序作为OH的氧化-到在pH〜11,而不管电极身份的水分子。在pH 5-10时,H +的扩散约束甚至在OER之前,通过氧化反应产生的氧化都会在电极表面附近产生pH〜4的环境,从而导致OER性能的整体pH无关区域。在这种无缓冲的接近中性pH的条件下,OER催化的同位素效应减弱,这与速率确定步骤(rds)是唯一的通过形成OO键,与质子传递解耦的电子传递步骤相符。 。尽管水分子是相同的反应物,但该反应机理不同于在更酸性条件下(pH <4)的反应机理。在酸性条件下,可观察到明显的同位素效应,这与在未配位的裸Ir位点上OO键的形成是最丰富的表面物种相一致。


























 京公网安备 11010802027423号
京公网安备 11010802027423号