当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Understanding the luminescence properties of Cu(I) complexes: a quantum chemical perusal
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2020-09-16 , DOI: 10.1039/d0cp04654j Nora Lüdtke 1, 2, 3, 4 , Jelena Föller 1, 2, 3, 4 , Christel M. Marian 1, 2, 3, 4
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2020-09-16 , DOI: 10.1039/d0cp04654j Nora Lüdtke 1, 2, 3, 4 , Jelena Föller 1, 2, 3, 4 , Christel M. Marian 1, 2, 3, 4
Affiliation
|
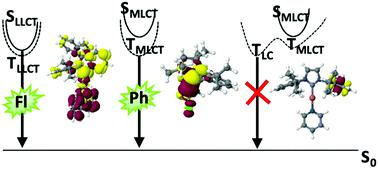
Electronic structures and excited-state properties of Cu(I) complexes with varying coordination numbers have been investigated by means of advanced quantum chemical methods. The computational protocol employs density functional-based methods for geometry optimizations and vibrational analyses including solvent effects through continuum models. Excitation energies, spin–orbit couplings and luminescence properties are evaluated using multireference configuration interaction methods. Rate constants of spin-allowed and spin-forbidden transitions have been determined according to the Fermi golden rule. The computational results for the 4-coordinate (DPEPhos)Cu(PyrTet), the 3-coordinate [IPr–Cu–Py2]+, and the linear CAACMe2–Cu–Cl complexes agree well with experimental absorption and emission wavelengths, intersystem crossing (ISC) time constants, and radiative lifetimes in liquid solution. Spectral shifts on the ligand-to-ligand charge transfer (LLCT) and metal-to-ligand charge transfer (MLCT) transitions caused by the polarity of the environment are well represented by the continuum models whereas the shifts caused by pseudo-Jahn–Teller distortions in the MLCT states are too pronounced in comparison to solid-state data. Systematic variation of the ligands in linear Cu(I) carbene complexes shows that only those complexes with S1 and T1 states of LLCT character possess sufficiently small singlet–triplet energy gaps ΔEST to enable thermally activated delayed fluorescence (TADF). Complexes whose S1 and T1 wavefunctions are dominated by MLCT excitations tend to emit phosphorescence instead. Unlike the situation in metal-free TADF emitters, the presence of low-lying locally excited triplet states does not promote ISC. These states rather hold the danger of trapping the excitation with nonradiative deactivation being the major deactivation channel.
中文翻译:

了解Cu(I)配合物的发光特性:量子化学研究
通过先进的量子化学方法研究了具有不同配位数的Cu(I)配合物的电子结构和激发态性质。该计算协议采用基于密度泛函的方法进行几何优化和振动分析,包括通过连续模型的溶剂影响。激发能,自旋轨道耦合和发光特性使用多参考配置相互作用方法进行评估。根据费米黄金法则确定了自旋允许和自旋禁止跃迁的速率常数。4坐标(DPEPhos)Cu(PyrTet),3坐标[IPr–Cu–Py 2 ] +和线性CAAC Me 2的计算结果–Cu–Cl络合物与实验吸收和发射波长,系统间穿越(ISC)时间常数以及液体溶液的辐射寿命非常吻合。连续模型很好地表示了由环境极性引起的配体到配体电荷转移(LLCT)和金属到配体电荷转移(MLCT)跃迁的光谱位移,而伪Jahn–Teller引起的位移与固态数据相比,MLCT状态中的失真太明显。线性Cu(I)卡宾配合物中配体的系统变化表明,只有那些具有LLCT特征的S 1和T 1状态的配合物具有足够小的单重态-三重态能隙ΔE ST以启用热激活延迟荧光(TADF)。S 1和T 1波函数受MLCT激发支配的复合物倾向于发出磷光。与无金属TADF发射器的情况不同,低位局部激发三重态的存在不会促进ISC。这些状态反而具有以非辐射失活作为主要失活通道捕获激发的危险。
更新日期:2020-10-19
中文翻译:
了解Cu(I)配合物的发光特性:量子化学研究
通过先进的量子化学方法研究了具有不同配位数的Cu(I)配合物的电子结构和激发态性质。该计算协议采用基于密度泛函的方法进行几何优化和振动分析,包括通过连续模型的溶剂影响。激发能,自旋轨道耦合和发光特性使用多参考配置相互作用方法进行评估。根据费米黄金法则确定了自旋允许和自旋禁止跃迁的速率常数。4坐标(DPEPhos)Cu(PyrTet),3坐标[IPr–Cu–Py 2 ] +和线性CAAC Me 2的计算结果–Cu–Cl络合物与实验吸收和发射波长,系统间穿越(ISC)时间常数以及液体溶液的辐射寿命非常吻合。连续模型很好地表示了由环境极性引起的配体到配体电荷转移(LLCT)和金属到配体电荷转移(MLCT)跃迁的光谱位移,而伪Jahn–Teller引起的位移与固态数据相比,MLCT状态中的失真太明显。线性Cu(I)卡宾配合物中配体的系统变化表明,只有那些具有LLCT特征的S 1和T 1状态的配合物具有足够小的单重态-三重态能隙ΔE ST以启用热激活延迟荧光(TADF)。S 1和T 1波函数受MLCT激发支配的复合物倾向于发出磷光。与无金属TADF发射器的情况不同,低位局部激发三重态的存在不会促进ISC。这些状态反而具有以非辐射失活作为主要失活通道捕获激发的危险。










































 京公网安备 11010802027423号
京公网安备 11010802027423号