当前位置:
X-MOL 学术
›
Anal. Chim. Acta
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
A graph density-based strategy for features fusion from different peak extract software to achieve more metabolites in metabolic profiling from high-resolution mass spectrometry
Analytica Chimica Acta ( IF 5.7 ) Pub Date : 2020-12-01 , DOI: 10.1016/j.aca.2020.09.029 Ran Ju , Xinyu Liu , Fujian Zheng , Xinjie Zhao , Xin Lu , Xiaohui Lin , Zhongda Zeng , Guowang Xu
Analytica Chimica Acta ( IF 5.7 ) Pub Date : 2020-12-01 , DOI: 10.1016/j.aca.2020.09.029 Ran Ju , Xinyu Liu , Fujian Zheng , Xinjie Zhao , Xin Lu , Xiaohui Lin , Zhongda Zeng , Guowang Xu
|
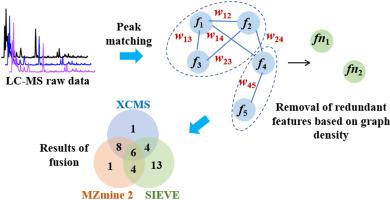
In metabolomics study, it is not easy to extract the metabolites from data of ultra high-performance liquid chromatography-high-resolution mass spectrometry, especially for those with low abundance. Different software for peak recognition and matching use different algorithms, leading to different extract results. Therefore, integration of results from different software can obtain richer metabolome information, but the redundant features should be removed. In this study, an integrated strategy of fusing features and removing redundancy based on graph density (FRRGD) was proposed. A graph is used to cover the ion features generated by two open access software (XCMS, MZmine 2) and a software (SIEVE) from an instrument vendor, and redundant features were removed by searching the maximal complete sub-graphs. A standard mixture containing 41 metabolites and a spontaneous urine were utilized to develop the method and demonstrate its usefulness. For the standard mixture, 19, 19 and 27 metabolites were extracted by XCMS, MZmine 2 and SIEVE, respectively. After fusion by FRRGD, 37 metabolites were obtained. For the diluted spontaneous urine sample, 1103, 1500 and 387 metabolites were extracted by XCMS, MZmine 2 and SIEVE, respectively, FRRGD produced 1619 metabolites which were much more than individual software, significantly increasing metabolome coverage. The proposed FRRGD shows a great prospect as a new data processing strategy for metabolomics study.
中文翻译:

一种基于图密度的策略,用于来自不同峰提取软件的特征融合,以在高分辨率质谱的代谢分析中获得更多代谢物
在代谢组学研究中,从超高效液相色谱-高分辨质谱数据中提取代谢物并不容易,尤其是对于低丰度的代谢物。不同的峰识别和匹配软件使用不同的算法,导致不同的提取结果。因此,整合不同软件的结果可以获得更丰富的代谢组信息,但应去除冗余特征。在这项研究中,提出了一种基于图密度(FRRGD)的融合特征和去除冗余的集成策略。一个图用于覆盖由两个开放访问软件(XCMS、MZmine 2)和一个仪器供应商的软件(SIEVE)生成的离子特征,并通过搜索最大完整子图去除冗余特征。使用包含 41 种代谢物和自发尿的标准混合物开发该方法并证明其有用性。对于标准混合物,分别通过 XCMS、MZmine 2 和 SIEVE 提取了 19、19 和 27 种代谢物。通过 FRRGD 融合后,获得 37 种代谢物。对于稀释的自发尿样本,XCMS、MZmine 2 和 SIEVE 分别提取了 1103、1500 和 387 种代谢物,FRRGD 产生了 1619 种代谢物,远远超过单个软件,显着提高了代谢组覆盖率。拟议的 FRRGD 作为代谢组学研究的新数据处理策略显示出巨大的前景。通过 FRRGD 融合后,获得 37 种代谢物。对于稀释的自发尿样本,XCMS、MZmine 2 和 SIEVE 分别提取了 1103、1500 和 387 种代谢物,FRRGD 产生了 1619 种代谢物,远远超过单个软件,显着提高了代谢组覆盖率。拟议的 FRRGD 作为代谢组学研究的新数据处理策略显示出巨大的前景。通过 FRRGD 融合后,获得 37 种代谢物。对于稀释的自发尿样本,XCMS、MZmine 2 和 SIEVE 分别提取了 1103、1500 和 387 种代谢物,FRRGD 产生了 1619 种代谢物,远远超过单个软件,显着提高了代谢组覆盖率。拟议的 FRRGD 作为代谢组学研究的新数据处理策略显示出巨大的前景。
更新日期:2020-12-01
中文翻译:
一种基于图密度的策略,用于来自不同峰提取软件的特征融合,以在高分辨率质谱的代谢分析中获得更多代谢物
在代谢组学研究中,从超高效液相色谱-高分辨质谱数据中提取代谢物并不容易,尤其是对于低丰度的代谢物。不同的峰识别和匹配软件使用不同的算法,导致不同的提取结果。因此,整合不同软件的结果可以获得更丰富的代谢组信息,但应去除冗余特征。在这项研究中,提出了一种基于图密度(FRRGD)的融合特征和去除冗余的集成策略。一个图用于覆盖由两个开放访问软件(XCMS、MZmine 2)和一个仪器供应商的软件(SIEVE)生成的离子特征,并通过搜索最大完整子图去除冗余特征。使用包含 41 种代谢物和自发尿的标准混合物开发该方法并证明其有用性。对于标准混合物,分别通过 XCMS、MZmine 2 和 SIEVE 提取了 19、19 和 27 种代谢物。通过 FRRGD 融合后,获得 37 种代谢物。对于稀释的自发尿样本,XCMS、MZmine 2 和 SIEVE 分别提取了 1103、1500 和 387 种代谢物,FRRGD 产生了 1619 种代谢物,远远超过单个软件,显着提高了代谢组覆盖率。拟议的 FRRGD 作为代谢组学研究的新数据处理策略显示出巨大的前景。通过 FRRGD 融合后,获得 37 种代谢物。对于稀释的自发尿样本,XCMS、MZmine 2 和 SIEVE 分别提取了 1103、1500 和 387 种代谢物,FRRGD 产生了 1619 种代谢物,远远超过单个软件,显着提高了代谢组覆盖率。拟议的 FRRGD 作为代谢组学研究的新数据处理策略显示出巨大的前景。通过 FRRGD 融合后,获得 37 种代谢物。对于稀释的自发尿样本,XCMS、MZmine 2 和 SIEVE 分别提取了 1103、1500 和 387 种代谢物,FRRGD 产生了 1619 种代谢物,远远超过单个软件,显着提高了代谢组覆盖率。拟议的 FRRGD 作为代谢组学研究的新数据处理策略显示出巨大的前景。










































 京公网安备 11010802027423号
京公网安备 11010802027423号