当前位置:
X-MOL 学术
›
Biochemistry
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Rational Design of Monodispersed Mutants of Proteins by Identifying Aggregation Contact Sites Using Solubilizing Agents.
Biochemistry ( IF 2.9 ) Pub Date : 2020-09-15 , DOI: 10.1021/acs.biochem.0c00414 Tatsuichiro Tsuji 1 , Sosuke Yoshinaga 1 , Mitsuhiro Takeda 1 , Takafumi Sato 1 , Akihiro Sonoda 1 , Norihito Ishida 1 , Kaori Yunoki 1 , Etsuko Toda 2, 3 , Yuya Terashima 2 , Kouji Matsushima 2 , Hiroaki Terasawa 1
Biochemistry ( IF 2.9 ) Pub Date : 2020-09-15 , DOI: 10.1021/acs.biochem.0c00414 Tatsuichiro Tsuji 1 , Sosuke Yoshinaga 1 , Mitsuhiro Takeda 1 , Takafumi Sato 1 , Akihiro Sonoda 1 , Norihito Ishida 1 , Kaori Yunoki 1 , Etsuko Toda 2, 3 , Yuya Terashima 2 , Kouji Matsushima 2 , Hiroaki Terasawa 1
Affiliation
|
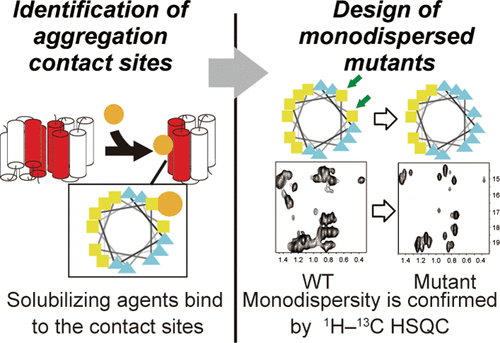
Suppression of protein aggregation is a subject of growing importance in the treatment of protein aggregation diseases, an urgent worldwide human health problem, and the production of therapeutic proteins, such as antibody drugs. We previously reported a method to identify compounds that suppress aggregation, based on screening using multiple terminal deletion mutants. We now present a method to determine the aggregation contact sites of proteins, using such solubilizing compounds, to design monodispersed mutants. We applied this strategy to the chemokine receptor-binding domain (CRBD) of FROUNT, which binds to the membrane-proximal C-terminal intracellular region of CCR2. Initially, the backbone NMR signals were assigned to a certain extent by available methods, and the putative locations of five α-helices were identified. Based on NMR chemical shift perturbations upon varying the protein concentrations, the first and third helices were found to contain the aggregation contact sites. The two helices are amphiphilic, and based on an NMR titration with 1,6-hexanediol, a CRBD solubilizing compound, the contact sites were identified as the hydrophobic patches located on the hydrophilic sides of the two helices. Subsequently, we designed multiple mutants targeting amino acid residues on the contact sites. Based on their NMR spectra, a doubly mutated CRBD (L538E/P612S) was selected from the designed mutants, and its monodispersed nature was confirmed by other biophysical methods. We then assessed the CCR2-binding activities of the mutants. Our method is useful for the protein structural analyses, the treatment of protein aggregation diseases, and the improvement of therapeutic proteins.
中文翻译:

通过使用增溶剂确定聚集接触位点,合理设计蛋白质的单分散突变体。
蛋白质聚集的抑制在蛋白质聚集疾病的治疗,全球范围内紧急的人类健康问题以及治疗性蛋白质如抗体药物的生产中日益重要。我们先前报道了一种基于使用多个末端缺失突变体进行筛选的方法来鉴定抑制聚集的化合物的方法。现在,我们提出一种使用这种增溶化合物来确定蛋白的聚集接触位点的方法,以设计单分散的突变体。我们将此策略应用于FROUNT的趋化因子受体结合域(CRBD),它与CCR2的膜近端C末端胞内区域结合。最初,通过可用方法在一定程度上分配了主链NMR信号,并确定了5个α螺旋的推定位置。基于改变蛋白质浓度后的NMR化学位移扰动,发现第一个和第三个螺旋包含聚集接触位点。这两个螺旋是两亲的,并且基于用1,6-己二醇(一种CRBD增溶化合物)的NMR滴定,接触点被确定为位于两个螺旋亲水侧的疏水斑点。随后,我们设计了针对接触位点上氨基酸残基的多个突变体。根据他们的NMR光谱,从设计的突变体中选择了双突变的CRBD(L538E / P612S),并通过其他生物物理方法证实了其单分散性。然后,我们评估了突变体的CCR2结合活性。我们的方法可用于蛋白质结构分析,蛋白质聚集疾病的治疗,
更新日期:2020-10-06
中文翻译:
通过使用增溶剂确定聚集接触位点,合理设计蛋白质的单分散突变体。
蛋白质聚集的抑制在蛋白质聚集疾病的治疗,全球范围内紧急的人类健康问题以及治疗性蛋白质如抗体药物的生产中日益重要。我们先前报道了一种基于使用多个末端缺失突变体进行筛选的方法来鉴定抑制聚集的化合物的方法。现在,我们提出一种使用这种增溶化合物来确定蛋白的聚集接触位点的方法,以设计单分散的突变体。我们将此策略应用于FROUNT的趋化因子受体结合域(CRBD),它与CCR2的膜近端C末端胞内区域结合。最初,通过可用方法在一定程度上分配了主链NMR信号,并确定了5个α螺旋的推定位置。基于改变蛋白质浓度后的NMR化学位移扰动,发现第一个和第三个螺旋包含聚集接触位点。这两个螺旋是两亲的,并且基于用1,6-己二醇(一种CRBD增溶化合物)的NMR滴定,接触点被确定为位于两个螺旋亲水侧的疏水斑点。随后,我们设计了针对接触位点上氨基酸残基的多个突变体。根据他们的NMR光谱,从设计的突变体中选择了双突变的CRBD(L538E / P612S),并通过其他生物物理方法证实了其单分散性。然后,我们评估了突变体的CCR2结合活性。我们的方法可用于蛋白质结构分析,蛋白质聚集疾病的治疗,










































 京公网安备 11010802027423号
京公网安备 11010802027423号