Journal of Proteomics ( IF 3.3 ) Pub Date : 2020-09-14 , DOI: 10.1016/j.jprot.2020.103986 Samantha Presslee 1 , Kirsty Penkman 2 , Roman Fischer 3 , Eden Richards-Slidel 4 , John Southon 5 , Carolina Acosta Hospitaleche 6 , Matthew Collins 7 , Ross MacPhee 8
|
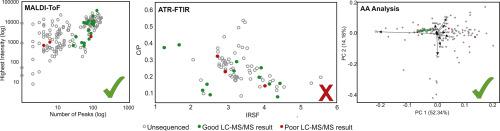
Ancient proteomics is being applied to samples dating further and further back in time, with many palaeontological specimens providing protein sequence data for phylogenetic analysis as well as protein degradation studies. However, fossils are a precious material and proteomic analysis is destructive and costly. In this paper we consider three different techniques (ATR-FTIR, MALDI-ToF MS and chiral AA analysis) to screen fossil material for potential protein preservation, aiming to maximise the proteomic information recovered and saving costly time consuming analyses which may produce low quality results. It was found that splitting factor and C/P indices from ATR-FTIR were not a reliable indicator of protein survival as they are confounded by secondary mineralisation of the fossil material. Both MALDI-ToF MS and chiral AA analysis results were able to successfully identify samples with surviving proteins, and it is suggested that one or both of these analyses be used for screening palaeontological specimens.
Significance
This study has shown both chiral amino acid analysis and MALDI-ToF MS are reliable screening methods for predicting protein survival in fossils. Both these methods are quick, cheap, minimally destructive (1 mg and 15 mg respectively) and can provide crucial additional information about the endogeneity of the surviving proteins. It is hoped that the use of these screening methods will encourage the examination of a wide range of palaeontological specimens for potential proteomic analysis. This in turn will give us a better understanding of protein survival far back in time and under different environmental conditions.
中文翻译:
评估选择古生物学骨样本进行肽测序的不同筛选方法。
远古的蛋白质组学被应用于年代久远的样品,许多古生物学标本提供蛋白质序列数据用于系统发育分析和蛋白质降解研究。但是,化石是一种宝贵的材料,蛋白质组学分析具有破坏性且成本高昂。在本文中,我们考虑了三种不同的技术(ATR-FTIR,MALDI-ToF MS和手性AA分析)来筛选化石材料以进行潜在的蛋白质保存,旨在最大程度地回收蛋白质组信息并节省可能产生低质量结果的昂贵耗时的分析。已经发现,ATR-FTIR的分裂因子和C / P指数不是蛋白质存活的可靠指标,因为它们与化石物质的二次矿化混淆了。
意义
这项研究表明,手性氨基酸分析和MALDI-ToF MS都是预测化石中蛋白质存活的可靠筛选方法。这两种方法都是快速,廉价,破坏性最小(分别为1 mg和15 mg),并且可以提供有关存活蛋白内源性的关键附加信息。希望使用这些筛选方法将鼓励检查各种古生物学标本,以进行潜在的蛋白质组学分析。反过来,这可以使我们更好地了解蛋白质在很久以前和在不同环境条件下的存活率。


























 京公网安备 11010802027423号
京公网安备 11010802027423号