当前位置:
X-MOL 学术
›
ACS Chem. Neurosci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Conventional molecular dynamics and metadynamics simulation studies of the binding and unbinding mechanism of TTR stabilizers AG10 and tafamidis.
ACS Chemical Neuroscience ( IF 5 ) Pub Date : 2020-09-11 , DOI: 10.1021/acschemneuro.0c00338 Shuangyan Zhou 1 , Siyu Ge 1 , Wenying Zhang 1 , Qiyuan Zhang 1 , Shuai Yuan 1 , Glenn V Lo 2 , Yusheng Dou 2
ACS Chemical Neuroscience ( IF 5 ) Pub Date : 2020-09-11 , DOI: 10.1021/acschemneuro.0c00338 Shuangyan Zhou 1 , Siyu Ge 1 , Wenying Zhang 1 , Qiyuan Zhang 1 , Shuai Yuan 1 , Glenn V Lo 2 , Yusheng Dou 2
Affiliation
|
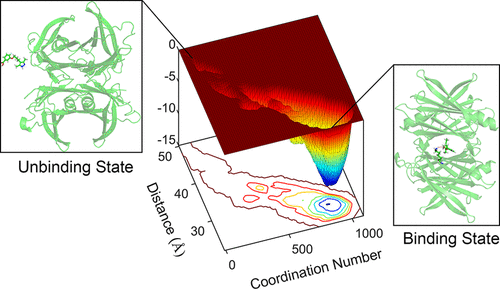
Amyloid transthyretin (ATTR) amyloidosis is a widespread and fatal systemic amyloidosis characterized by the misfolding and amyloid aggregation of transthyretin (TTR). Studies suggest that dissociation of the TTR tetramer is the key step for its misfolding. Because of the importance of tetramer dissociation on ATTR amyloidosis, many TTR stabilizers have been discovered to stabilize the tetramer structure. This paper describes the application conventional molecular dynamics and metadynamics simulations to investigate the binding and unbinding mechanisms of two TTR stabilizers, including AG10 and tafamidis. AG10 has been granted an orphan drug designation by the U.S. Food and Drug Administration (FDA), and tafamidis was the first FDA-approved treatment for ATTR cardiomyopathy. The conventional molecular dynamics simulations reveal that both AG10 and tafamidis can stabilize the TTR tetramer through different mechanisms. AG10 stabilizes TTR tetramer by forming H-bonds with S117 to mimic the protective effect of T119M. Tafamidis stabilizes the tetramer by forming H-bond with S52 in the flexible CD loop to increase its structural stability. Despite the strong binding affinity of tafamidis, the free-energy surface constructed from metadynamics simulation suggests that tafamidis unbinds more readily than AG10 with lower free-energy barriers between the binding state and other intermediates. Finally, by performing pharmacophore analysis, we found two common important moieties of the studied compounds for their binding on the pockets, which can provide valuable guidance for future lead compounds’ optimization in designing drugs for ATTR amyloidosis.
中文翻译:

TTR稳定剂AG10和塔法米第的结合和解离机理的常规分子动力学和元动力学模拟研究。
淀粉样运甲状腺素蛋白(ATTR)淀粉样变性病是一种广泛而致命的全身性淀粉样变性病,其特征在于运甲状腺素蛋白(TTR)的错误折叠和淀粉样蛋白聚集。研究表明,TTR四聚体的解离是其错误折叠的关键步骤。由于四聚体解离对ATTR淀粉样变性的重要性,已发现许多TTR稳定剂可稳定四聚体结构。本文介绍了应用常规分子动力学和元动力学模拟来研究两种TTR稳定剂(包括AG10和塔法米第)的结合和未结合机理。AG10已被美国食品和药物管理局(FDA)授予孤儿药称号,而塔法米迪是FDA批准的第一种治疗ATTR心肌病的药物。常规的分子动力学模拟表明,AG10和塔法米迪都可以通过不同的机理稳定TTR四聚体。AG10通过与S117形成H键来模拟T119M的保护作用,从而稳定了TTR四聚体。Tafamidis通过在柔性CD环中与S52形成H键来稳定四聚体,从而增加其结构稳定性。尽管塔法米迪具有很强的结合亲和力,但通过代谢动力学模拟构建的自由能表面表明,与结合状态较弱的AG10相比,塔法米迪更易于解离,其结合态与其他中间体之间的自由能垒较低。最后,通过进行药效基团分析,我们发现了所研究化合物的两个共同的重要部分,即它们在口袋上的结合,
更新日期:2020-10-07
中文翻译:
TTR稳定剂AG10和塔法米第的结合和解离机理的常规分子动力学和元动力学模拟研究。
淀粉样运甲状腺素蛋白(ATTR)淀粉样变性病是一种广泛而致命的全身性淀粉样变性病,其特征在于运甲状腺素蛋白(TTR)的错误折叠和淀粉样蛋白聚集。研究表明,TTR四聚体的解离是其错误折叠的关键步骤。由于四聚体解离对ATTR淀粉样变性的重要性,已发现许多TTR稳定剂可稳定四聚体结构。本文介绍了应用常规分子动力学和元动力学模拟来研究两种TTR稳定剂(包括AG10和塔法米第)的结合和未结合机理。AG10已被美国食品和药物管理局(FDA)授予孤儿药称号,而塔法米迪是FDA批准的第一种治疗ATTR心肌病的药物。常规的分子动力学模拟表明,AG10和塔法米迪都可以通过不同的机理稳定TTR四聚体。AG10通过与S117形成H键来模拟T119M的保护作用,从而稳定了TTR四聚体。Tafamidis通过在柔性CD环中与S52形成H键来稳定四聚体,从而增加其结构稳定性。尽管塔法米迪具有很强的结合亲和力,但通过代谢动力学模拟构建的自由能表面表明,与结合状态较弱的AG10相比,塔法米迪更易于解离,其结合态与其他中间体之间的自由能垒较低。最后,通过进行药效基团分析,我们发现了所研究化合物的两个共同的重要部分,即它们在口袋上的结合,


























 京公网安备 11010802027423号
京公网安备 11010802027423号