Bioorganic Chemistry ( IF 4.5 ) Pub Date : 2020-09-12 , DOI: 10.1016/j.bioorg.2020.104274 Mennatallah A. Shaheen , Ali A. El-Emam , Nadia S. El-Gohary
|
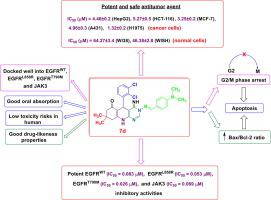
New series of hexahydroquinoline and fused quinoline derivatives were designed and synthesized. The thirty seven new compounds were screened for in vitro antitumor activity against HepG2, HCT-116 and MCF-7 cancer cells. Results indicated that compounds 2e, 2h, 5b, 5c, 6a, 7d and 9b have the strongest potency against the three cancer cells, and they were further screened for in vitro cytotoxicity against A431 and H1975 cancer cells, as well as WI38 and WISH normal cells. Results revealed that 7d potently inhibited the growth of H1975 cells harboring EGFRT790M mutation (IC50 = 1.32 ± 0.2 µM) over A431 cells overexpressing EGFRWT (IC50 = 4.96 ± 0.3 µM). Moreover, the seven compounds displayed low cytotoxicity against the tested normal cells. The seven potent antitumor compounds were examined for their ability to inhibit the activity of EGFRWT. The attained data manifested that 7d has remarkable EGFRWT inhibitory activity (IC50 = 0.083 ± 0.002 μM) compared to erlotinib (IC50 = 0.067 ± 0.002 μM). Compound 7d was further studied for its enzymatic inhibitory activity against other eight human kinases, and it displayed outstanding inhibitory activity against EGFRL858R and EGFRT790M mutants (IC50 = 0.053 ± 0.002, 0.026 ± 0.001 μM, respectively), as well as JAK3 (IC50 = 0.069 ± 0.003 μM). Analysis of cell cycle evidenced that 7d induces cell cycle arrest in G2/M and pre-G1 phases in the tested cancer cells. In addition, cancer cell death induced by 7d was proved to take place via apoptosis supported by elevated Bax/Bcl-2 ratio in the tested cancer cells. Moreover, docking results confirmed the good binding interactions of 7d with EGFRWT, EGFRL858R, EGFRT790M and JAK3, which came in agreement with the results of in vitro enzyme assay. Further, 7d is predicted to have good oral absorption, good drug-likeness properties and low toxicity risks in human.
中文翻译:
新系列六氢喹啉和稠合喹啉衍生物作为野生型EGFR和突变EGFR(L858R和T790M)的有效抑制剂的设计,合成和生物学评估
设计并合成了一系列新的六氢喹啉和稠合喹啉衍生物。筛选了三十七个新化合物对HepG2,HCT-116和MCF-7癌细胞的体外抗肿瘤活性。结果表明化合物2e,2h,5b,5c,6a,7d和9b对这三种癌细胞具有最强的效力,并且进一步筛选了它们对A431和H1975癌细胞以及WI38和WISH正常细胞的体外细胞毒性细胞。结果显示,与 过表达EGFR WT(IC )的A431细胞相比,7d有效抑制了带有EGFR T790M突变(IC 50 = 1.32±0.2 µM)的H1975细胞的生长50 = 4.96±0.3 µM)。此外,这七个化合物对测试的正常细胞显示出低细胞毒性。检查了七种有效的抗肿瘤化合物抑制EGFR WT活性的能力。获得的数据表明, 与厄洛替尼(IC 50 = 0.067± 0.002μM)相比,7d具有显着的EGFR WT抑制活性(IC 50 = 0.083± 0.002μM)。进一步研究了化合物7d对其他8种人类激酶的酶促抑制活性,并显示出对EGFR L858R和EGFR T790M突变体的出色抑制活性(IC 50 分别为0.053±0.002、0.026±0.001μM)和JAK3(IC 50 = 0.069±0.003μM)。细胞周期分析表明,7d诱导受试癌细胞的G2 / M和pre-G1期细胞周期停滞。另外,在被测试的癌细胞中,由7d诱导的癌细胞死亡被证明是通过凋亡的发生而实现的,该凋亡由升高的Bax / Bcl-2比支持。此外,对接结果证实7d与EGFR WT,EGFR L858R,EGFR T790M和JAK3具有良好的结合相互作用,这与体外酶测定的结果一致。此外,第7天 预计对人体具有良好的口服吸收,相似的药物特性和低毒性风险。










































 京公网安备 11010802027423号
京公网安备 11010802027423号