当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
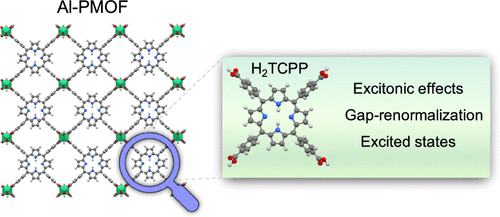
From Isolated Porphyrin Ligands to Periodic Al-PMOF: A Comparative Study of the Optical Properties Using DFT/TDDFT
The Journal of Physical Chemistry C ( IF 3.7 ) Pub Date : 2020-09-10 , DOI: 10.1021/acs.jpcc.0c06885 Andres Ortega-Guerrero 1 , Maria Fumanal 1 , Gloria Capano 1 , Berend Smit 1
The Journal of Physical Chemistry C ( IF 3.7 ) Pub Date : 2020-09-10 , DOI: 10.1021/acs.jpcc.0c06885 Andres Ortega-Guerrero 1 , Maria Fumanal 1 , Gloria Capano 1 , Berend Smit 1
Affiliation
|
The use of photosensitizers as organic ligands in metal–organic frameworks (MOFs) is a common practice to engineer their UV/vis optical absorption. For instance, MOFs consisting of porphyrin ligands usually inherit their light-harvesting properties and thus follow the Gouterman model in which the low-lying excitations correspond to π → π* transitions. However, the characterization of the excited states of porphyrin ligands in MOFs requires an appropriate description of the periodic crystal including the metal nodes and intermolecular interactions. Here, we investigate the UV/vis absorption properties of porphyrin-MOF Al-PMOF, its two metalated forms, Zn-Al-PMOF and Co-Al-PMOF, and their corresponding isolated porphyrin ligands using density functional theory (DFT)/time-dependent DFT (TDDFT) simulations with Perdew–Burke–Ernzerhof (PBE), PBE0, and CAM-B3LYP functionals. Our results indicate that hybrid functionals are necessary to capture the proper nature of the transitions and the excitonic effects of the optical and fundamental gaps in porphyrin molecules and porphyrin-MOFs that the PBE functional fails to describe. Likewise, the simulations show that a wrong representation of some excitations can be obtained depending on the functional and when the Tamm–Dancoff approximation is used. Finally, our results show that the PBE and PBE0 functionals are not able to capture the gap renormalization when going from the isolated molecules to the periodic crystals. Overall, the nature of the optical transitions, excitonic effects, and gap renormalization are important features to assess in the prediction of optical properties in MOF crystals that require considering proper functionals and approximations to overcome the main failures of DFT/TDDFT calculations.
中文翻译:

从孤立的卟啉配体到周期性的Al-PMOF:使用DFT / TDDFT光学性质的比较研究
在金属有机框架(MOF)中使用光敏剂作为有机配体是设计其UV / vis光学吸收的常用方法。例如,由卟啉配体组成的MOF通常继承其光捕获特性,因此遵循Gouterman模型,其中低洼的激发对应于π→π*跃迁。但是,MOF中卟啉配体的激发态的表征需要对包括金属节点和分子间相互作用的周期晶体进行适当的描述。在这里,我们使用密度泛函理论(DFT)/时间研究卟啉-MOF Al-PMOF,其两种金属化形式Zn-Al-PMOF和Co-Al-PMOF及其对应的分离卟啉配体的UV / vis吸收特性。 Perdew–Burke–Ernzerhof(PBE),PBE0的DFT(TDDFT)相关模拟,和CAM-B3LYP功能。我们的结果表明,杂化功能对于捕获过渡的正确性质以及卟啉分子和卟啉-MOF中光学和基本间隙的激子效应是必要的,而PBE功能无法描述这些杂化。同样,仿真表明,根据函数和使用Tamm–Dancoff近似值时,可以获得一些激励的错误表示。最后,我们的结果表明,当从分离的分子进入周期性晶体时,PBE和PBE0的功能无法捕获间隙重新归一化。总体而言,光学跃迁的性质,激子效应,
更新日期:2020-10-02
中文翻译:
从孤立的卟啉配体到周期性的Al-PMOF:使用DFT / TDDFT光学性质的比较研究
在金属有机框架(MOF)中使用光敏剂作为有机配体是设计其UV / vis光学吸收的常用方法。例如,由卟啉配体组成的MOF通常继承其光捕获特性,因此遵循Gouterman模型,其中低洼的激发对应于π→π*跃迁。但是,MOF中卟啉配体的激发态的表征需要对包括金属节点和分子间相互作用的周期晶体进行适当的描述。在这里,我们使用密度泛函理论(DFT)/时间研究卟啉-MOF Al-PMOF,其两种金属化形式Zn-Al-PMOF和Co-Al-PMOF及其对应的分离卟啉配体的UV / vis吸收特性。 Perdew–Burke–Ernzerhof(PBE),PBE0的DFT(TDDFT)相关模拟,和CAM-B3LYP功能。我们的结果表明,杂化功能对于捕获过渡的正确性质以及卟啉分子和卟啉-MOF中光学和基本间隙的激子效应是必要的,而PBE功能无法描述这些杂化。同样,仿真表明,根据函数和使用Tamm–Dancoff近似值时,可以获得一些激励的错误表示。最后,我们的结果表明,当从分离的分子进入周期性晶体时,PBE和PBE0的功能无法捕获间隙重新归一化。总体而言,光学跃迁的性质,激子效应,


























 京公网安备 11010802027423号
京公网安备 11010802027423号