当前位置:
X-MOL 学术
›
ChemPhysChem
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Structural and photophysical properties of various polypyridyl ligands: A combined experimental and computational study.
ChemPhysChem ( IF 2.9 ) Pub Date : 2020-09-11 , DOI: 10.1002/cphc.202000592 Liesbeth De Bruecker 1 , Jonas Everaert 2 , Pascal Van Der Voort 3 , Christian V Stevens 2 , Michel Waroquier 1 , Veronique Van Speybroeck 1
ChemPhysChem ( IF 2.9 ) Pub Date : 2020-09-11 , DOI: 10.1002/cphc.202000592 Liesbeth De Bruecker 1 , Jonas Everaert 2 , Pascal Van Der Voort 3 , Christian V Stevens 2 , Michel Waroquier 1 , Veronique Van Speybroeck 1
Affiliation
|
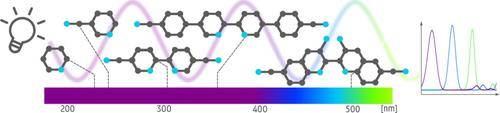
Covalent triazine frameworks (CTFs) with polypyridyl ligands are very promising supports to anchor photocatalytic complexes. Herein, we investigate the photophysical properties of a series of ligands which vary by the extent of the aromatic system, the nitrogen content and their topologies to aid in selecting interesting building blocks for CTFs. Interestingly, some linkers have a rotational degree of freedom, allowing both a trans and cis structure, where only the latter allows anchoring. Therefore, the influence of the dihedral angle on the UV‐Vis spectrum is studied. The photophysical properties are investigated by a combined computational and experimental study. Theoretically, both static and molecular dynamics simulations are performed to deduce ground‐ and excited state properties based on density functional theory (DFT) and time‐dependent DFT. The position of the main absorption peak shifts towards higher wavelengths for an increased size of the π‐system and a higher π‐electron deficiency. We found that the position of the main absorption peak among the different ligands studied in this work can amount to 271 nm; which has a significant impact on the photophysical properties of the ligands. This broad range of shifts allows modulation of the electronic structure by varying the ligands and may help in a rational design of efficient photocatalysts.
中文翻译:

各种聚吡啶配体的结构和光物理性质:实验和计算相结合的研究。
具有聚吡啶基配体的共价三嗪框架(CTF)是非常有前途的锚定光催化复合物的载体。在这里,我们研究了一系列配体的光物理性质,这些配体随芳香系统的范围、氮含量及其拓扑而变化,以帮助选择有趣的 CTF 构建块。有趣的是,一些接头具有旋转自由度,允许反式和顺式结构,其中只有后者允许锚定。因此,研究了二面角对紫外-可见光谱的影响。通过计算和实验相结合的研究来研究光物理性质。理论上,静态和分子动力学模拟都是基于密度泛函理论 (DFT) 和瞬态 DFT 来推断基态和激发态性质。由于π系统尺寸的增加和更高的π电子缺陷,主吸收峰的位置向更高的波长移动。我们发现本工作研究的不同配体的主吸收峰位置可达271 nm;这对配体的光物理性质有显着影响。这种广泛的变化允许通过改变配体来调节电子结构,并可能有助于高效光催化剂的合理设计。
更新日期:2020-11-18
中文翻译:
各种聚吡啶配体的结构和光物理性质:实验和计算相结合的研究。
具有聚吡啶基配体的共价三嗪框架(CTF)是非常有前途的锚定光催化复合物的载体。在这里,我们研究了一系列配体的光物理性质,这些配体随芳香系统的范围、氮含量及其拓扑而变化,以帮助选择有趣的 CTF 构建块。有趣的是,一些接头具有旋转自由度,允许反式和顺式结构,其中只有后者允许锚定。因此,研究了二面角对紫外-可见光谱的影响。通过计算和实验相结合的研究来研究光物理性质。理论上,静态和分子动力学模拟都是基于密度泛函理论 (DFT) 和瞬态 DFT 来推断基态和激发态性质。由于π系统尺寸的增加和更高的π电子缺陷,主吸收峰的位置向更高的波长移动。我们发现本工作研究的不同配体的主吸收峰位置可达271 nm;这对配体的光物理性质有显着影响。这种广泛的变化允许通过改变配体来调节电子结构,并可能有助于高效光催化剂的合理设计。


























 京公网安备 11010802027423号
京公网安备 11010802027423号