Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy ( IF 4.3 ) Pub Date : 2020-09-11 , DOI: 10.1016/j.saa.2020.118935 Yi-Wei Fan , Huai-Qian Wang , Hui-Fang Li
|
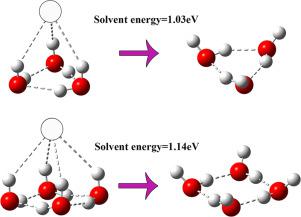
The hydrated clusters (n = 1–4) in gas phase are studied by density functional theory calculations (DFT) coupled with stochastic kicking method. The global minimum structure of exhibits low-symmetry pattern since only one H atom of water molecules interact with Co− ion and other ones associate with a network of hydrogen bonds. The Co− ion prefers to locate at vertex site of the water molecular clusters in such way to reduce the repulsion with O atom. These results elucidate the formation of these low-lying isomers are determined by the delicate balance between ion-water and water-water interactions.
中文翻译:
的Co Microsolvation -在水:加上随机踢方法密度泛函理论计算
水合簇 通过密度泛函理论计算(DFT)与随机踢法相结合,研究了气相(n = 1-4)。的全球最小结构因为交互水分子的仅一个H原子表现出低的对称图案钴-离子和其他的关联与氢键的网络。所述钴-离子倾向于在以这样的方式的水分子簇的顶点部位来定位,以减少与氧原子的排斥力。这些结果阐明了这些低洼异构体的形成是由离子-水与水-水相互作用之间的微妙平衡决定的。









































 京公网安备 11010802027423号
京公网安备 11010802027423号