当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Ionization Energy and Reduction Potential in Ferrocene Derivatives: Comparison of Hybrid and Pure DFT Functionals.
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2020-09-08 , DOI: 10.1021/acs.jpca.0c06663 Mateja Toma 1 , Tea Kuvek 1 , Valerije Vrček 1
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2020-09-08 , DOI: 10.1021/acs.jpca.0c06663 Mateja Toma 1 , Tea Kuvek 1 , Valerije Vrček 1
Affiliation
|
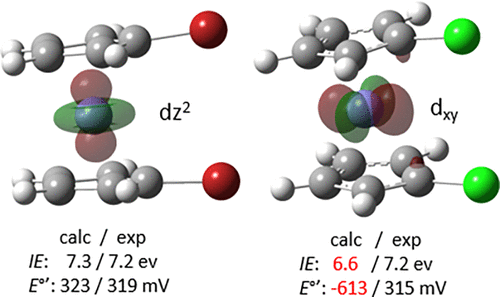
Hybrid density functionals have been regularly applied in state-of-the-art computational models for predicting reduction potentials. Benchmark calculations of the absolute reduction potential of ferricenium/ferrocene couple, the IUPAC-proposed reference in nonaqueous solution, include the B3LYP/6-31G(d)/LanL2TZf protocol. We used this procedure to calculate ionization energies and reduction potentials for a comprehensive set of ferrocene derivatives. The protocol works very well for a number of derivatives. However, a significant discrepancy (>1 V) between experimental and calculated data was detected for selected cases. Three variables were assessed to detect an origin of the observed failure: density functional, basis set, and solvation model. It comes out that the Hartree–Fock exchange fraction in hybrid-DFT methods is the main source of the error. The accidental errors were observed for other hybrid models like PBE0, BHandHLYP, and M06-2X. Therefore, hybrid DFT methods should be used with caution, or pure functionals (BLYP or M06L) may be used instead.
中文翻译:

二茂铁衍生物的电离能和还原电位:杂化和纯DFT功能的比较。
混合密度函数已定期应用于最新的计算模型中,以预测还原电位。IUPAC建议的非水溶液参考中的铁/二茂铁对绝对还原电位的基准计算包括B3LYP / 6-31G(d)/ LanL2TZf方案。我们使用此程序来计算一套完整的二茂铁衍生物的电离能和还原电位。该协议对许多派生协议非常有效。但是,在某些情况下,实验数据与计算数据之间存在明显差异(> 1 V)。评估了三个变量以检测观察到的故障的根源:密度泛函,基集和溶剂化模型。结果表明,混合DFT方法中的Hartree-Fock交换分数是误差的主要来源。对于其他混合模型(如PBE0,BHandHLYP和M06-2X),观察到了意外错误。因此,应谨慎使用混合DFT方法,或者可以使用纯功能(BLYP或M06L)代替。
更新日期:2020-10-02
中文翻译:
二茂铁衍生物的电离能和还原电位:杂化和纯DFT功能的比较。
混合密度函数已定期应用于最新的计算模型中,以预测还原电位。IUPAC建议的非水溶液参考中的铁/二茂铁对绝对还原电位的基准计算包括B3LYP / 6-31G(d)/ LanL2TZf方案。我们使用此程序来计算一套完整的二茂铁衍生物的电离能和还原电位。该协议对许多派生协议非常有效。但是,在某些情况下,实验数据与计算数据之间存在明显差异(> 1 V)。评估了三个变量以检测观察到的故障的根源:密度泛函,基集和溶剂化模型。结果表明,混合DFT方法中的Hartree-Fock交换分数是误差的主要来源。对于其他混合模型(如PBE0,BHandHLYP和M06-2X),观察到了意外错误。因此,应谨慎使用混合DFT方法,或者可以使用纯功能(BLYP或M06L)代替。










































 京公网安备 11010802027423号
京公网安备 11010802027423号