当前位置:
X-MOL 学术
›
Int. J. Quantum Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Structural, electronic, and thermodynamic properties of TiO2/organic clusters: performance of DFTB method with different parameter sets
International Journal of Quantum Chemistry ( IF 2.3 ) Pub Date : 2020-09-09 , DOI: 10.1002/qua.26427 Vladimir S. Naumov 1, 2 , Anastasiia S. Loginova 1 , Alexander A. Avdoshin 1 , Stanislav K. Ignatov 1 , Alexey V. Mayorov 3, 4 , Bálint Aradi 2 , Thomas Frauenheim 2
International Journal of Quantum Chemistry ( IF 2.3 ) Pub Date : 2020-09-09 , DOI: 10.1002/qua.26427 Vladimir S. Naumov 1, 2 , Anastasiia S. Loginova 1 , Alexander A. Avdoshin 1 , Stanislav K. Ignatov 1 , Alexey V. Mayorov 3, 4 , Bálint Aradi 2 , Thomas Frauenheim 2
Affiliation
|
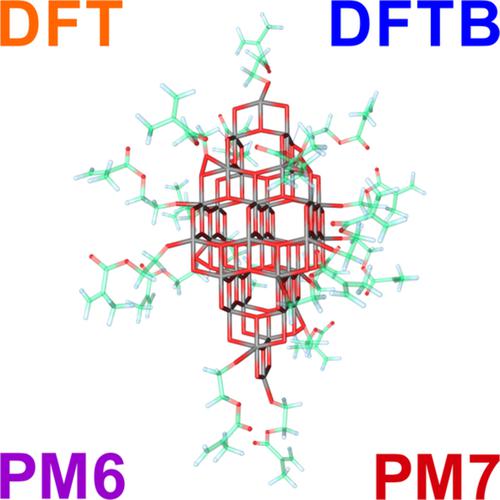
The clusters of bare TiO2 and TiO2 with linked organic ligands modeling polyorganic composites used as photocatalytic materials were studied using the density functional based tight binding (DFTB) electronic structure method with three parameter sets (trans3d, tiorg, and matsci) in comparison with results of B3LYP/6‐31G(d,p) calculations, semiempirical methods PM6 and PM7, and available experimental data. It was found that the highly scalable DFTB method shows results that are close to the B3LYP/6‐31G(d,p) level of theory. The corrected version of the tiorg DFTB parameter set (tiorg‐smooth) has better performance for the estimations of structural parameters, whereas the trans3d set better reproduces energies of the composite material formation in polycondensation reactions. Performance of the matsci set is somehow in the middle of the tiorg‐smooth and trans3d sets. The tiorg‐smooth and matsci sets can be used for the studies of adsorption complexes of bare TiO2 clusters. All three DFTB parameter sets well estimate the electronic parameters of clusters (HOMO‐LUMO gap, ionization potential, and dipole moment). DFTB results are closer to the estimates made with DFT (B3LYP/6‐31G(d,p)) than the results of PM6 and PM7 methods. DFTB calculations of large (up to 448 atoms) bare TiO2 and TiO2/organic clusters (72 structures in total) confirm the robustness and computational efficiency of the method.
中文翻译:

TiO2 /有机团簇的结构,电子和热力学性质:具有不同参数集的DFTB方法的性能
使用基于密度泛函的紧密结合(DFTB)电子结构方法和三个参数集(trans3d,tiorg和matsci),研究了裸露的TiO 2和TiO 2与连接的有机配体的簇状结构,该簇模拟了用作光催化材料的多有机复合材料。 B3LYP / 6-31G(d,p)计算的结果,PM6和PM7的半经验方法以及可用的实验数据。结果发现,高度可扩展的DFTB方法显示的结果接近理论的B3LYP / 6-31G(d,p)水平。tiorg DFTB参数集的更正版本(tiorg-smooth)具有更好的结构参数估计性能,而trans3d设置可以更好地复制缩聚反应中复合材料形成的能量。Matsci集的性能在tiorg-smooth集和trans3d集之间处于某种程度上。所述tiorg光滑和matsci可用于裸露的TiO吸附复合物的研究组2集群。这三个DFTB参数集都很好地估计了簇的电子参数(HOMO-LUMO间隙,电离电势和偶极矩)。与PM6和PM7方法相比,DFTB结果更接近于DFT(B3LYP / 6-31G(d,p))所作的估计。大型(最多448个原子)裸TiO 2和TiO 2 /有机簇(总共72个结构)的DFTB计算证实了该方法的鲁棒性和计算效率。
更新日期:2020-09-09
中文翻译:
TiO2 /有机团簇的结构,电子和热力学性质:具有不同参数集的DFTB方法的性能
使用基于密度泛函的紧密结合(DFTB)电子结构方法和三个参数集(trans3d,tiorg和matsci),研究了裸露的TiO 2和TiO 2与连接的有机配体的簇状结构,该簇模拟了用作光催化材料的多有机复合材料。 B3LYP / 6-31G(d,p)计算的结果,PM6和PM7的半经验方法以及可用的实验数据。结果发现,高度可扩展的DFTB方法显示的结果接近理论的B3LYP / 6-31G(d,p)水平。tiorg DFTB参数集的更正版本(tiorg-smooth)具有更好的结构参数估计性能,而trans3d设置可以更好地复制缩聚反应中复合材料形成的能量。Matsci集的性能在tiorg-smooth集和trans3d集之间处于某种程度上。所述tiorg光滑和matsci可用于裸露的TiO吸附复合物的研究组2集群。这三个DFTB参数集都很好地估计了簇的电子参数(HOMO-LUMO间隙,电离电势和偶极矩)。与PM6和PM7方法相比,DFTB结果更接近于DFT(B3LYP / 6-31G(d,p))所作的估计。大型(最多448个原子)裸TiO 2和TiO 2 /有机簇(总共72个结构)的DFTB计算证实了该方法的鲁棒性和计算效率。










































 京公网安备 11010802027423号
京公网安备 11010802027423号